Uludağ Alkaya Dilek, Usluer Esra, Alp Ünkar Zeynep, Şeker Ali, Adaletli İbrahim, Güneş Nilay, Madazlı Rıza, Kadıoğlu Pınar, Derbent Murat, Tüysüz Beyhan
Department of Pediatric Genetics, Cerrahpaşa Medical Faculty, Istanbul University-Cerrahpaşa, Istanbul, Turkey.
Department of Neonatology, Cerrahpaşa Medical Faculty, Istanbul University-Cerrahpaşa, Istanbul, Turkey.
Calcif Tissue Int. 2025 Apr 8;116(1):59. doi: 10.1007/s00223-025-01366-w.
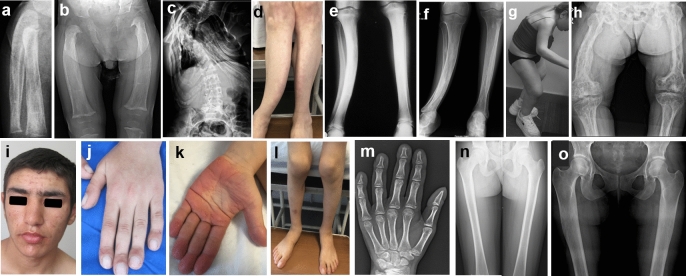
Osteosclerotic bone diseases include more than 30 rare diseases characterized by excessive bone formation. The aim of this study is to compare the molecular pathogenesis and prognostic features of 12 different osteosclerotic diseases. Thirty-four patients from 23 families were included, 25 of whom were followed for a period of one to 22 years. Exome sequencing was performed in 20 families. Primary hypertrophic osteoarthropathy (PHOAR1/2) was found in 12 patients, followed by juvenile Paget's disease (JPD)-5 in five, craniometaphyseal dysplasia (CMD) and Camurati-Engelmann disease (CED) in four, Ghosal hematodiaphyseal dysplasia (GHDD) in three patients, sclerosteosis-1 in two patients, and ultra-rare diseases including trichothiodystrophy-1, prenatal Caffey disease, melorheosteosis, and Lenz-Majewski hyperostotic dwarfism in one patient each. Patients with CMD and sclerosteosis-1 had severe cranial sclerosis leading to facial dysmorphism. CMD was characterized by metaphyseal widening, radiolucency, and diaphyseal sclerosis of the long bones in early childhood and later developed Erlenmeyer flask deformity sparing the vertebrae and pelvis, whereas sclerosteosis-1 manifested as generalized sclerosis. CED and GHDD share bone pain, difficulty in walking, and diaphyseal sclerosis, with some patients also having bone marrow involvement. Interestingly, patients with CED and JPD-5 showed osteopenia in early childhood, followed by the development of osteosclerosis in late childhood. Clinical and radiologic findings improved over time in PHOAR1 patients, whereas they progressed in JPD-5 and trichothiodystrophy-1 patients. Intra- and interfamilial clinical differences were observed in CMD, CED, JPD-5, and GHDD. The knowledge gained about the natural history of osteosclerotic diseases will make an important contribution to their diagnosis and management.
骨硬化性骨病包括30多种以骨形成过多为特征的罕见病。本研究的目的是比较12种不同骨硬化性疾病的分子发病机制和预后特征。纳入了来自23个家庭的34例患者,其中25例随访了1至22年。对20个家庭进行了外显子组测序。12例患者被诊断为原发性肥大性骨关节病(PHOAR1/2),其次是5例青少年佩吉特病(JPD)-5、4例颅骨骨干发育异常(CMD)和卡-恩病(CED)、3例戈萨尔骨干发育异常(GHDD)、2例骨硬化症-1,还有1例患者分别患有罕见病,包括毛发硫营养不良-1、产前卡菲病、肢骨纹状肥大和伦茨-马耶夫斯基骨肥大性侏儒症。CMD和骨硬化症-1患者有严重的颅骨硬化,导致面部畸形。CMD的特征是儿童早期长骨干骺端增宽、透亮区和骨干硬化,随后发展为烧瓶样畸形,不累及椎骨和骨盆,而骨硬化症-1表现为全身硬化。CED和GHDD都有骨痛、行走困难和骨干硬化,一些患者还伴有骨髓受累。有趣的是,CED和JPD-5患者在儿童早期表现为骨质减少,随后在儿童晚期发展为骨硬化。PHOAR1患者的临床和影像学表现随时间改善,而JPD-5和毛发硫营养不良-1患者的病情则进展。在CMD、CED、JPD-5和GHDD患者中观察到家族内和家族间的临床差异。所获得的关于骨硬化性疾病自然史的知识将对其诊断和管理做出重要贡献。