Guan Haoyue, Sun Huimin, Zhao Xia
National Institute for Food and Drug Control, Beijing 100050, China.
Int J Mol Sci. 2025 Apr 1;26(7):3262. doi: 10.3390/ijms26073262.
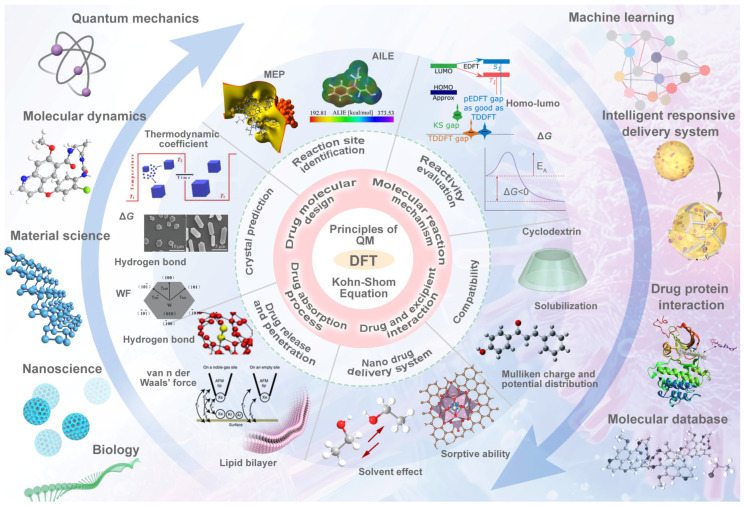
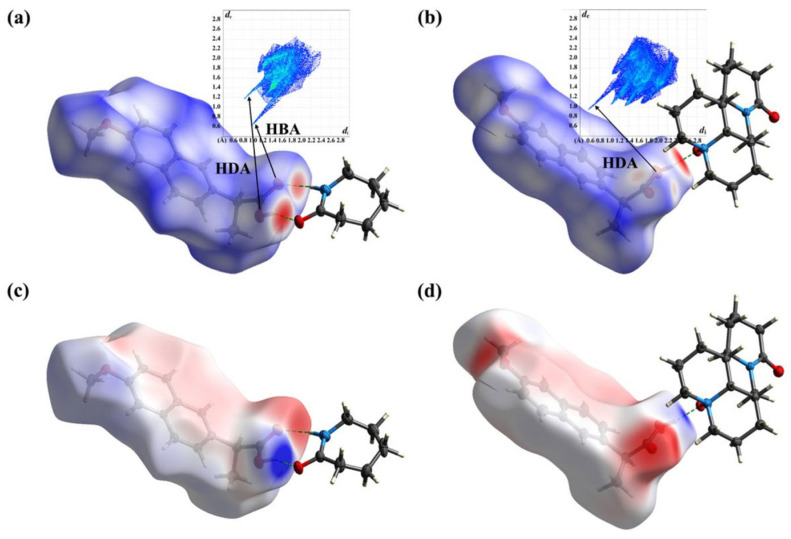
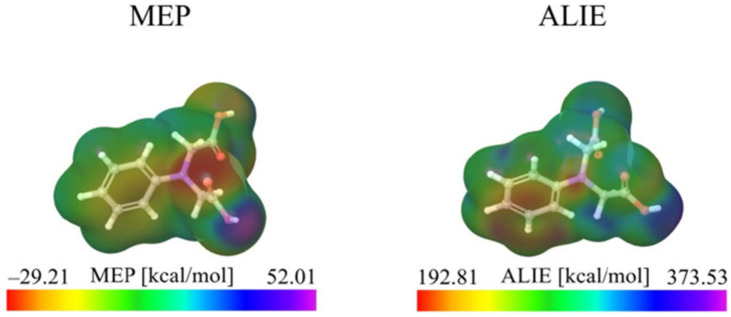
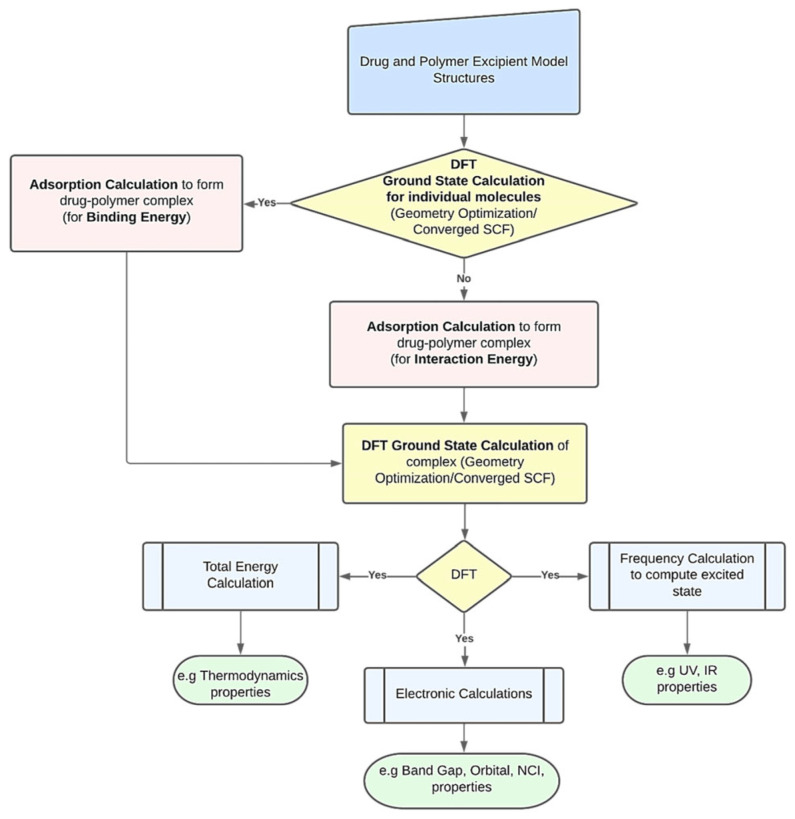
This review systematically examines the pivotal applications of the Density Functional Theory (DFT) in drug formulation design, emphasizing its capability to elucidate molecular interaction mechanisms through quantum mechanical calculations. By solving the Kohn-Sham equations with precision up to 0.1 kcal/mol, DFT enables accurate electronic structure reconstruction, providing theoretical guidance for optimizing drug-excipient composite systems. In solid dosage forms, DFT clarifies the electronic driving forces governing active pharmaceutical ingredient (API)-excipient co-crystallization, predicting reactive sites and guiding stability-oriented co-crystal design. For nanodelivery systems, DFT optimizes carrier surface charge distribution through van der Waals interactions and π-π stacking energy calculations, thereby enhancing targeting efficiency. Furthermore, DFT combined with solvation models (e.g., COSMO) quantitatively evaluates polar environmental effects on drug release kinetics, delivering critical thermodynamic parameters (e.g., ΔG) for controlled-release formulation development. Notably, DFT-driven co-crystal thermodynamic analysis and pH-responsive release mechanism modeling substantially reduce experimental validation cycles. While DFT faces challenges in dynamic simulations of complex solvent environments, its integration with molecular mechanics and multiscale frameworks has achieved computational breakthroughs. This work offers interdisciplinary methodology support for accelerating data-driven formulation design.
本综述系统地研究了密度泛函理论(DFT)在药物制剂设计中的关键应用,强调了其通过量子力学计算阐明分子相互作用机制的能力。通过精确求解Kohn-Sham方程,精度可达0.1 kcal/mol,DFT能够实现准确的电子结构重建,为优化药物-辅料复合体系提供理论指导。在固体剂型中,DFT阐明了控制活性药物成分(API)-辅料共结晶的电子驱动力,预测反应位点并指导面向稳定性的共晶设计。对于纳米递送系统,DFT通过范德华相互作用和π-π堆积能计算优化载体表面电荷分布,从而提高靶向效率。此外,DFT与溶剂化模型(如COSMO)相结合,定量评估极性环境对药物释放动力学的影响,为控释制剂开发提供关键的热力学参数(如ΔG)。值得注意的是,DFT驱动的共晶热力学分析和pH响应释放机制建模大大减少了实验验证周期。虽然DFT在复杂溶剂环境的动态模拟中面临挑战,但其与分子力学和多尺度框架的整合已取得计算上的突破。这项工作为加速数据驱动的制剂设计提供了跨学科的方法支持。