Das Anibh Martin
Department of Paediatric Kidney, Liver and Metabolic Diseases, Hannover Medical School, Hannover, Germany.
Hannover Medical School, Carl Neuberg Str. 1, D- 30625, Hannover, Germany.
Metab Brain Dis. 2025 Apr 26;40(5):192. doi: 10.1007/s11011-025-01619-5.
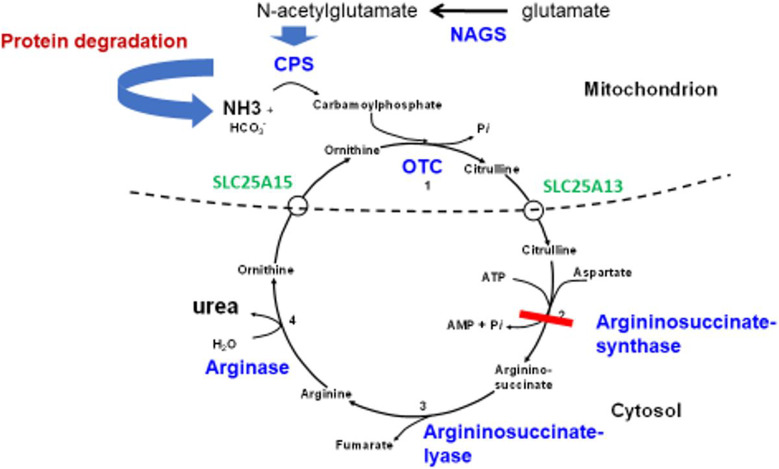
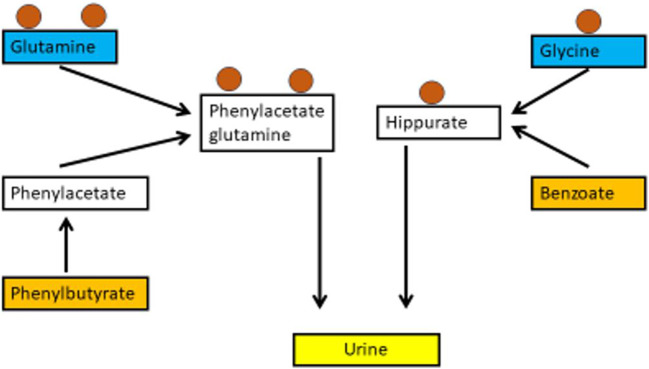
Hyperammonaemia is an important cause for encephalopathy. Ammonia is the waste product of amino acid degradation and cannot be excreted via urine. Ammonia is metabolized to water-soluble urea via the urea cycle. Hyperammonaemia not only occurs during acute liver failure, but also in rare genetically determined defects of enzymes or transporters involved in the urea cycle resulting in elevated ammonia concentrations. Enzyme defects include deficiency of carbamylphosphate synthase, N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate lyase and arginase, transporter defects are citrin deficiency and HHH-syndrome. These urea cycle defects (UCD) mostly manifest for the first time during the neonatal period, infancy or childhood, however first clinical manifestations including encephalopathy may be observed in adulthood in milder forms. Therefore, physicians treating adults should be aware of clinical symptoms in UCD to make a timely diagnosis and initiate treatment. In adulthood, clinical symptoms are often uncharacteristic including headache, avoidance of high-protein food, psychiatric symptoms triggered by heavy exercise or delivery of a child, autism, attention deficit, lethargy, developmental delay and epilepsy. Elevated ammonia concentrations in blood are the biochemical hallmark. Some UCDs can be diagnosed at metabolite level, others only at genetic level. Treatment consists of eucaloric, low-protein diet supplemented with essential amino acids and vitamins/trace elements, and intake of arginine or citrulline. Pharmacological scavengers of nitrogen are benzoate and butyrate. If conservative therapy fails, hemodialysis should be considered. Prompt treatment during acute crises is essential for optimal outcome. Liver transplantation is considered in metabolically unstable patients. For arginase deficiency, enzyme replacement therapy is available.
高氨血症是脑病的一个重要病因。氨是氨基酸降解的终产物,不能经尿液排出。氨通过尿素循环代谢为水溶性的尿素。高氨血症不仅发生在急性肝衰竭时,也可因参与尿素循环的酶或转运体罕见的基因缺陷导致氨浓度升高而引起。酶缺陷包括氨甲酰磷酸合成酶、N - 乙酰谷氨酸合成酶、鸟氨酸转氨甲酰酶、精氨琥珀酸裂解酶和精氨酸酶缺乏,转运体缺陷包括柠檬酸转运蛋白缺乏和HHH综合征。这些尿素循环缺陷(UCD)大多在新生儿期、婴儿期或儿童期首次出现临床表现,不过在成年期也可能以较轻的形式出现包括脑病在内的首发临床表现。因此,治疗成人患者的医生应了解UCD的临床症状以便及时诊断并开始治疗。在成年期,临床症状往往不典型,包括头痛、避免高蛋白食物、剧烈运动或分娩引发的精神症状、自闭症、注意力缺陷、嗜睡、发育迟缓及癫痫。血氨浓度升高是生化标志。一些UCD可在代谢物水平诊断,另一些则只能在基因水平诊断。治疗包括给予补充必需氨基酸及维生素/微量元素的等热量、低蛋白饮食,以及摄入精氨酸或瓜氨酸。氮的药理清除剂有苯甲酸盐和丁酸盐。如果保守治疗无效,应考虑血液透析。急性危象期间及时治疗对获得最佳疗效至关重要。对于代谢不稳定的患者,可考虑肝移植。对于精氨酸酶缺乏症,可采用酶替代疗法。