Smith Ewan W, Hamilton William L, Warne Ben, Walker Elena R, Jahun Aminu S, Hosmillo Myra, Gupta Ravindra K, Goodfellow Ian, Gkrania-Klotsas Effrossyni, Török M Estée, Illingworth Christopher J R
MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow, United Kingdom.
Department of Medicine, University of Cambridge, Cambridge, United Kingdom.
PLoS Pathog. 2025 Apr 28;21(4):e1013109. doi: 10.1371/journal.ppat.1013109. eCollection 2025 Apr.
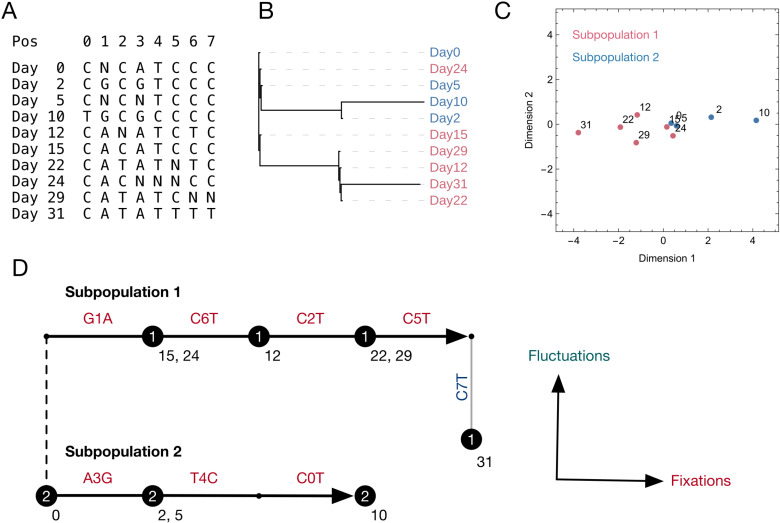
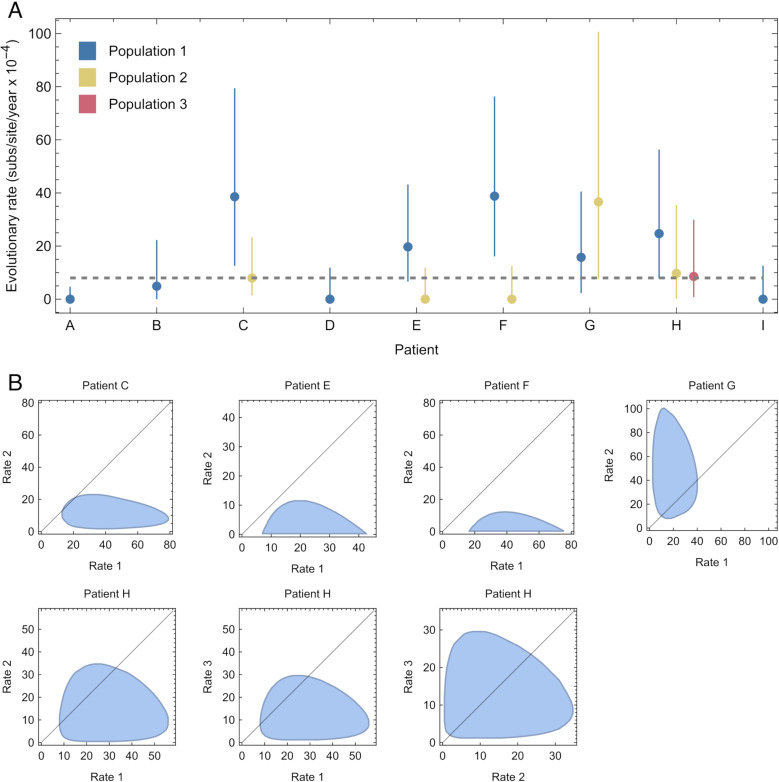
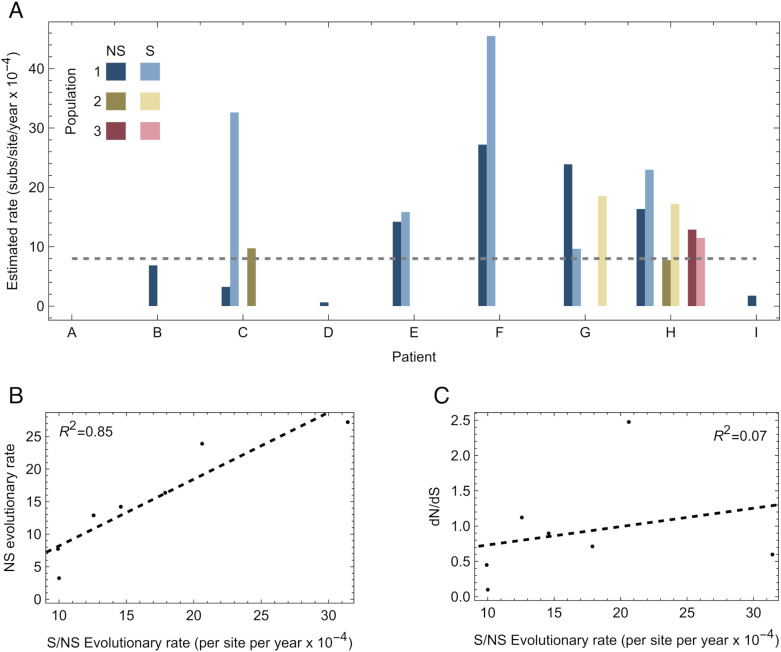
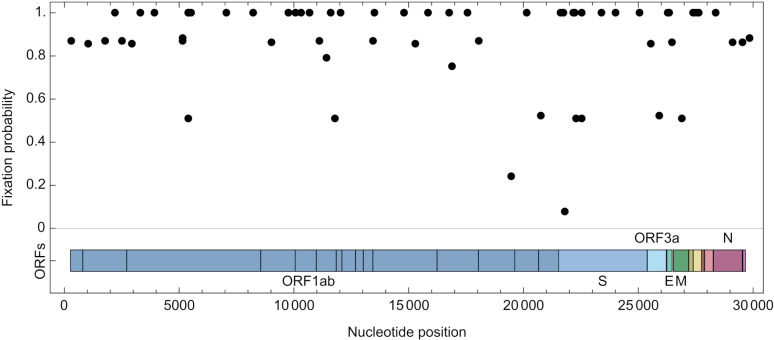
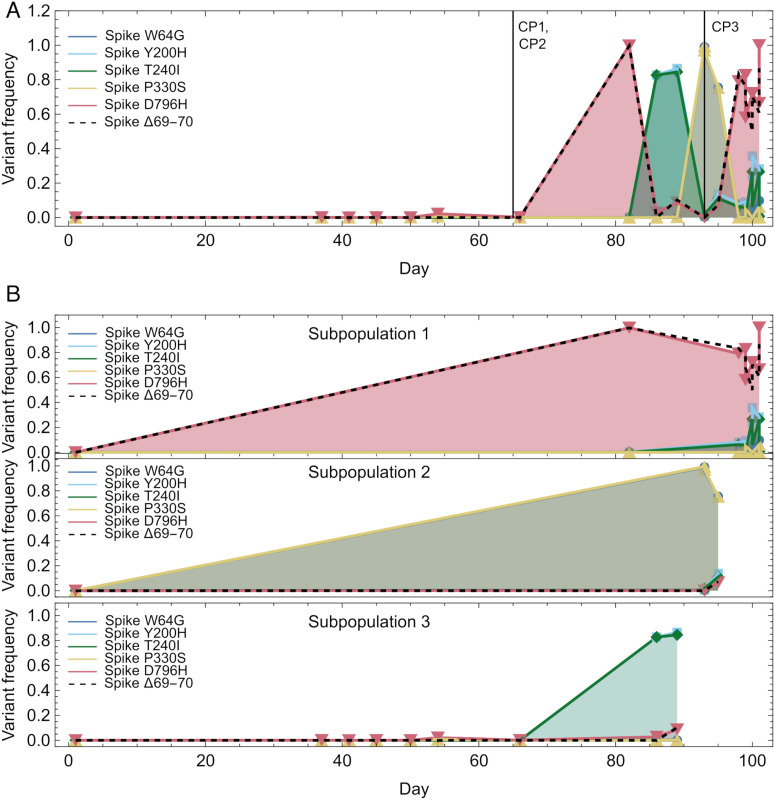
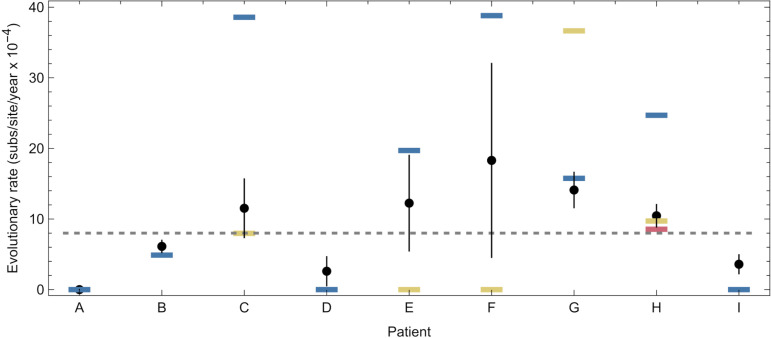
An important feature of the evolution of the SARS-CoV-2 virus has been the emergence of highly mutated novel variants, which are characterised by the gain of multiple mutations relative to viruses circulating in the general global population. Cases of chronic viral infection have been suggested as an explanation for this phenomenon, whereby an extended period of infection, with an increased rate of evolution, creates viruses with substantial genetic novelty. However, measuring a rate of evolution during chronic infection is made more difficult by the potential existence of compartmentalisation in the viral population, whereby the viruses in a host form distinct subpopulations. We here describe and apply a novel statistical method to study within-host virus evolution, identifying the minimum number of subpopulations required to explain sequence data observed from cases of chronic infection, and inferring rates for within-host viral evolution. Across nine cases of chronic SARS-CoV-2 infection in hospitalised patients we find that non-trivial population structure is relatively common, with five cases showing evidence of more than one viral population evolving independently within the host. The detection of non-trivial population structure was more common in severely immunocompromised individuals (p = 0.04, Fisher's Exact Test). We find cases of within-host evolution proceeding significantly faster, and significantly slower, than that of the global SARS-CoV-2 population, and of cases in which viral subpopulations in the same host have statistically distinguishable rates of evolution. Non-trivial population structure was associated with high rates of within-host evolution that were systematically underestimated by a more standard inference method.
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)病毒进化的一个重要特征是出现了高度变异的新型变体,其特征是相对于全球普通人群中传播的病毒而言获得了多个突变。慢性病毒感染病例被认为是这一现象的一个解释,即延长的感染期以及加快的进化速度产生了具有大量基因新异性的病毒。然而,由于病毒群体中可能存在区室化现象,即宿主中的病毒形成不同的亚群,这使得测量慢性感染期间的进化速度变得更加困难。我们在此描述并应用一种新颖的统计方法来研究宿主内病毒进化,确定解释从慢性感染病例中观察到的序列数据所需的最少亚群数量,并推断宿主内病毒进化的速率。在9例住院患者的慢性SARS-CoV-2感染病例中,我们发现非平凡的群体结构相对常见,有5例显示有多个病毒群体在宿主体内独立进化的证据。在严重免疫功能低下的个体中,检测到非平凡的群体结构更为常见(p = 0.04,Fisher精确检验)。我们发现宿主内进化的病例比全球SARS-CoV-2群体的进化速度显著更快和显著更慢,以及同一宿主内病毒亚群具有统计学上可区分的进化速率的病例。非平凡的群体结构与宿主内进化的高速度相关,而一种更标准的推断方法会系统性地低估这种高速度。