Liang Hewei, Zou Yuanqiang, Wang Mengmeng, Hu Tongyuan, Wang Haoyu, He Wenxin, Ju Yanmei, Guo Ruijin, Chen Junyi, Guo Fei, Zeng Tao, Dong Yuliang, Zhang Yuning, Wang Bo, Liu Chuanyu, Jin Xin, Zhang Wenwei, Xu Xun, Xiao Liang
BGI Research, Shenzhen 518083, China.
BGI Research, Wuhan 430074, China.
GigaByte. 2025 Apr 25;2025:gigabyte154. doi: 10.46471/gigabyte.154. eCollection 2025.
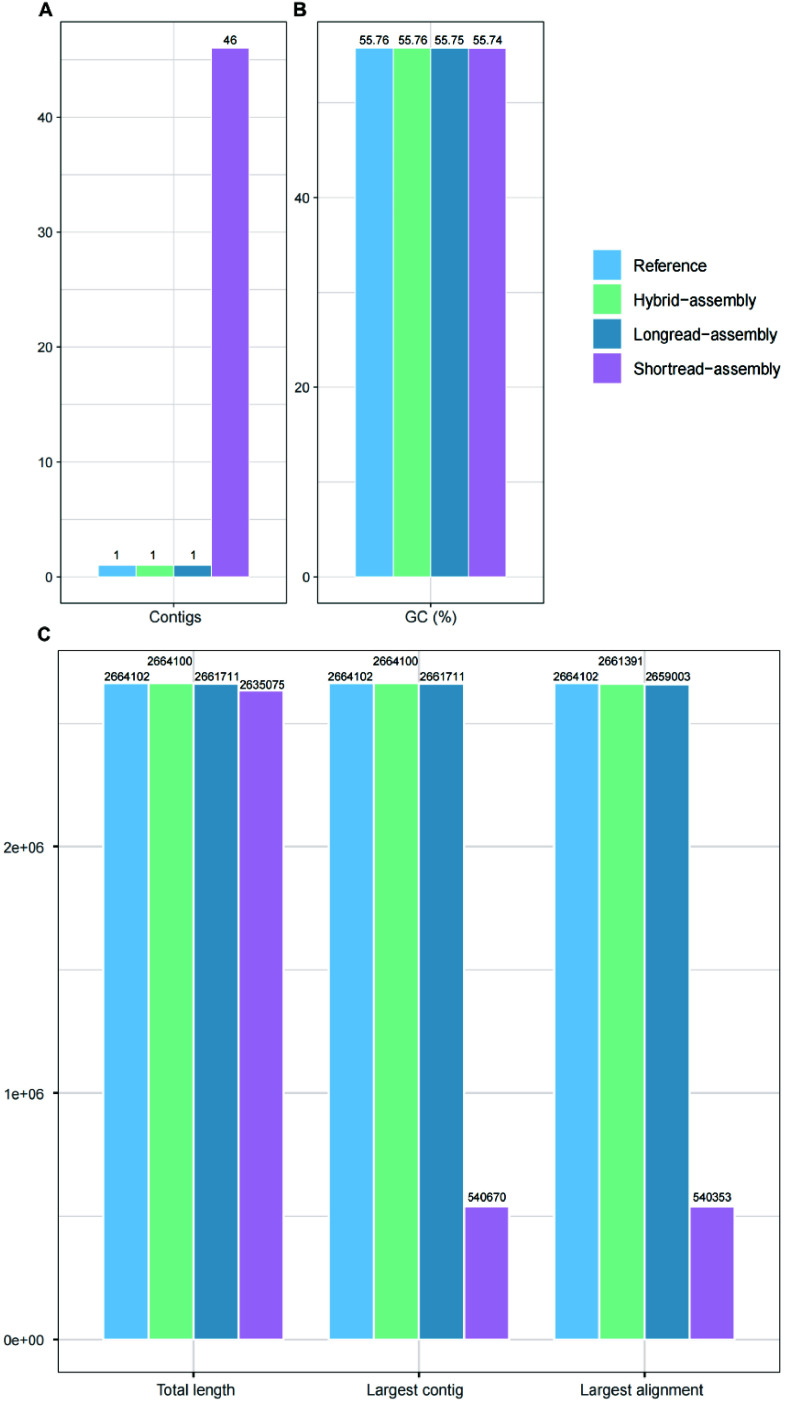
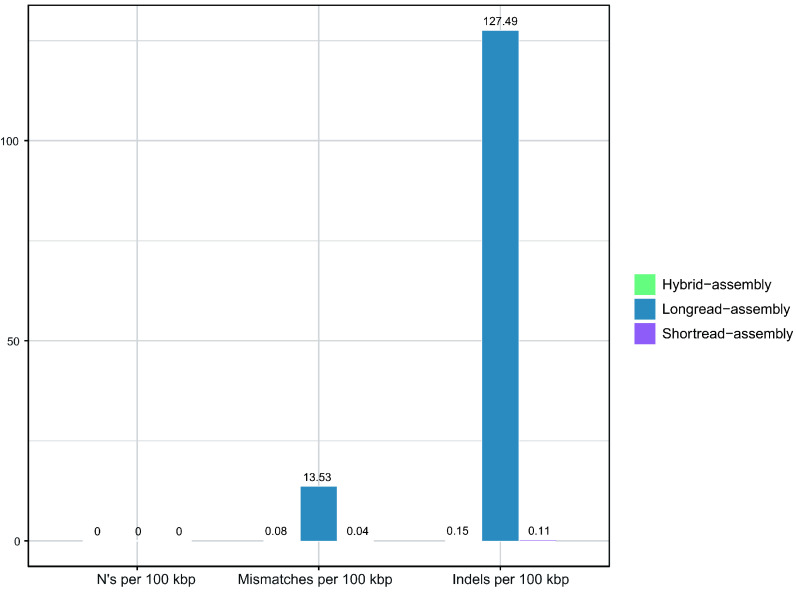
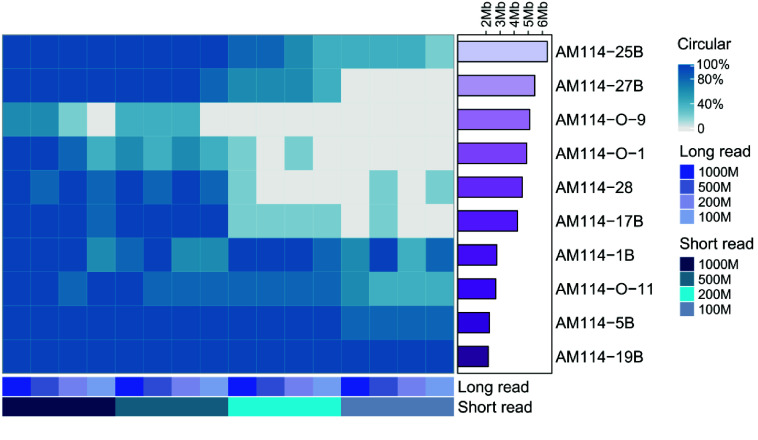
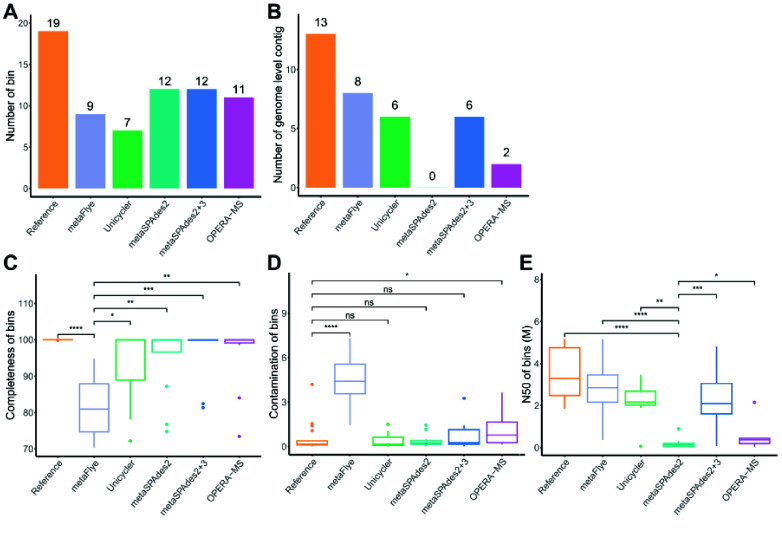
Current microbial sequencing relies on short-read platforms like Illumina and DNBSEQ, which are cost-effective and accurate but often produce fragmented draft genomes. Here, we used CycloneSEQ for long-read sequencing of ATCC BAA-835, producing long-reads with an average length of 11.6 kbp and an average quality score of 14.4. Hybrid assembly with short-reads data resulted in an error rate of only 0.04 mismatches and 0.08 indels per 100 kbp compared to the reference genome. This method, validated across nine species, successfully assembled complete circular genomes. Hybrid assembly significantly enhances genome completeness by using long-reads to fill gaps and accurately assembling multi-copy rRNA genes, unlike short-reads alone. Data subsampling showed that combining over 500 Mbp of short-read data with 100 Mbp of long-read data yields high-quality circular assemblies. CycloneSEQ long-reads improves the assembly of circular complete genomes from mixed microbial communities; however, its base quality needs improving. Integrating DNBSEQ short-reads improved accuracy, resulting in complete and accurate assemblies.
当前的微生物测序依赖于Illumina和DNBSEQ等短读长平台,这些平台具有成本效益且准确,但通常会产生碎片化的基因组草图。在这里,我们使用CycloneSEQ对ATCC BAA-835进行长读长测序,产生的长读长平均长度为11.6 kbp,平均质量分数为14.4。与短读长数据进行混合组装,与参考基因组相比,每100 kbp的错误率仅为0.04个错配和0.08个插入缺失。该方法在九个物种中得到验证,成功组装出完整的环状基因组。与仅使用短读长不同,混合组装通过使用长读长填补缺口并准确组装多拷贝rRNA基因,显著提高了基因组的完整性。数据二次抽样表明,将超过500 Mbp的短读长数据与100 Mbp的长读长数据相结合,可产生高质量的环状组装。CycloneSEQ长读长改进了来自混合微生物群落的环状完整基因组的组装;然而,其碱基质量需要改进。整合DNBSEQ短读长提高了准确性,从而实现了完整且准确的组装。