Ju Yi, Zhang Yuting, Tian Xiaolin, Zhu Nanbin, Zheng Yufan, Qiao Yiming, Yang Tao, Niu Baolin, Li Xiaoyun, Yu Liu, Liu Zhuolin, Wu Yixuan, Zhi Yang, Dong Yinuo, Xu Qingling, Yang Xiaoming, Wang Xuening, Wang Xiaokai, Deng Haiteng, Mao Yimin, Li Xiaobo
Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Fudan University, Shanghai, China.
MOE Key Laboratory of Bioinformatics, School of Life Sciences, Tsinghua University, Beijing, China.
Redox Biol. 2025 Jun;83:103660. doi: 10.1016/j.redox.2025.103660. Epub 2025 May 6.
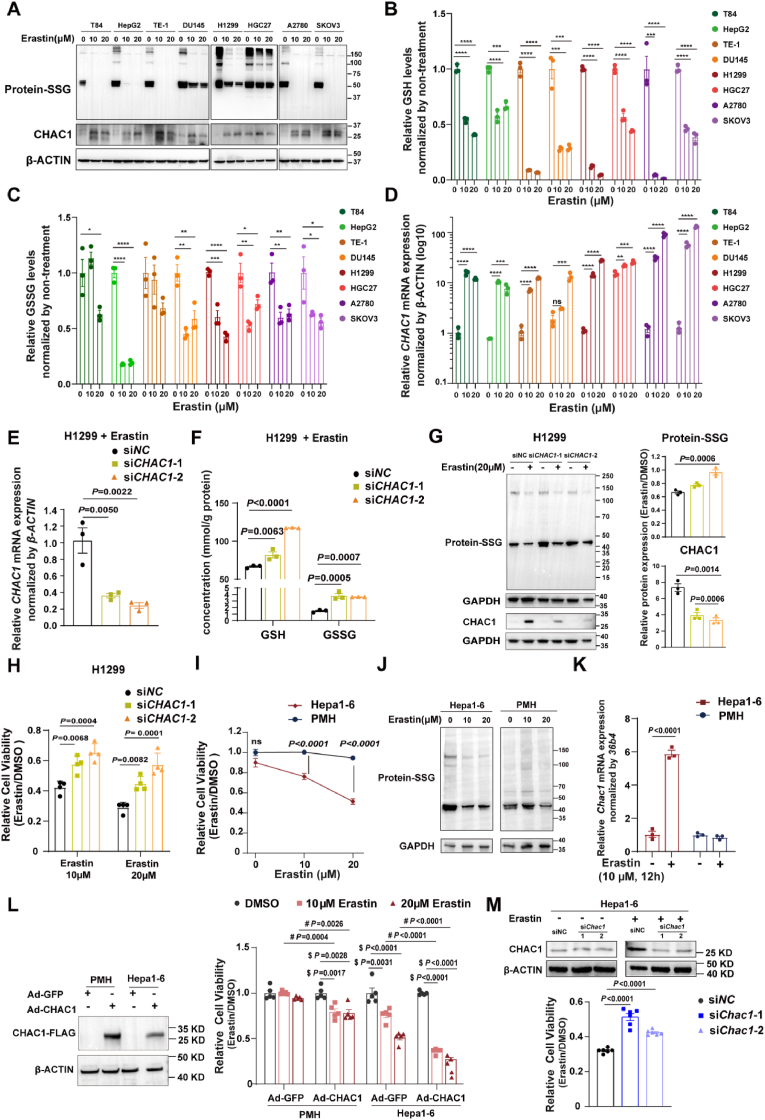
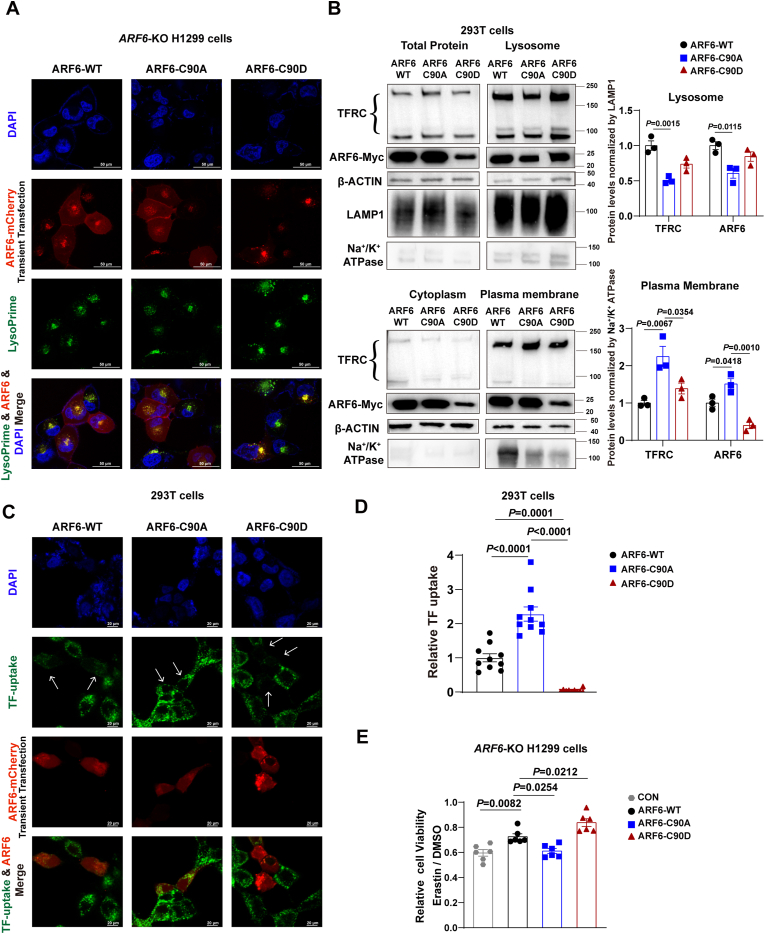
Ferroptosis is one of the most critical biological consequences of glutathione depletion. Excessive oxidative stress, indicated by an elevated oxidized glutathione (GSSG)/reduced glutathione (GSH) ratio, is recognized as a key driver of ferroptosis. However, in glutathione depletion-induced ferroptosis, a marked decrease in total glutathione levels (including both GSH and GSSG) is frequently observed, yet its significance remains understudied. Protein S-glutathionylation (protein-SSG) levels are closely linked to the redox state and cellular glutathione pools including GSH and GSSG. To date, the role of protein-SSG during cell ferroptosis induced by glutathione depletion remains poorly understood. Here, we demonstrated that upregulation of CHAC1, a glutathione-degrading enzyme, acted as a key regulator of protein-SSG formation and exacerbated glutathione depletion-induced ferroptosis. This effect was observed in both in vitro and in vivo models, including erastin-induced ferroptosis across multiple cell lines and acetaminophen overdose-triggered ferroptosis in hepatocytes. Deficiency of CHAC1 resulted in increased glutathione pools, enhanced protein-SSG, improved liver function, and attenuation of hepatocyte ferroptosis upon acetaminophen challenge. These protective effects were reversed by CHAC1 overexpression. Using quantitative redox proteomics, we identified glutathione pool-sensitive S-glutathionylated proteins. As an important example, we discovered that ADP-ribosylation factor 6 (ARF6) was regulated by S-glutathionylation during glutathione depletion-induced ferroptosis. Our findings revealed that CHAC1 upregulation reduced the S-glutathionylation of ARF6, resulting in decreased ARF6 levels in lysosomes. This, in turn, enhanced the localization of the transferrin receptor (TFRC) on the cell membrane and increased transferrin uptake, ultimately compromising the protective role of ARF6 in ferroptosis induced by glutathione depletion. Targeting TFRC using GalNAc-siTfrc mitigated acetaminophen-induced liver injury in vivo. In conclusion, our study provide evidence that availability of glutathione pools affects protein S-glutathionylation and regulates protein functions to influence the process of ferroptosis, which opens an avenue to understanding the cell ferroptosis induced by glutathione depletion.
铁死亡是谷胱甘肽耗竭最关键的生物学后果之一。氧化型谷胱甘肽(GSSG)/还原型谷胱甘肽(GSH)比值升高所表明的过度氧化应激被认为是铁死亡的关键驱动因素。然而,在谷胱甘肽耗竭诱导的铁死亡中,经常观察到总谷胱甘肽水平(包括GSH和GSSG)显著下降,但其意义仍未得到充分研究。蛋白质S-谷胱甘肽化(蛋白质-SSG)水平与氧化还原状态以及包括GSH和GSSG在内的细胞谷胱甘肽池密切相关。迄今为止,蛋白质-SSG在谷胱甘肽耗竭诱导的细胞铁死亡中的作用仍知之甚少。在此,我们证明了谷胱甘肽降解酶CHAC1的上调是蛋白质-SSG形成的关键调节因子,并加剧了谷胱甘肽耗竭诱导的铁死亡。在体外和体内模型中均观察到了这种效应,包括多种细胞系中erastin诱导的铁死亡以及对乙酰氨基酚过量引发的肝细胞铁死亡。CHAC1缺陷导致谷胱甘肽池增加、蛋白质-SSG增强、肝功能改善以及对乙酰氨基酚攻击后肝细胞铁死亡的减轻。这些保护作用被CHAC1过表达所逆转。使用定量氧化还原蛋白质组学,我们鉴定了对谷胱甘肽池敏感的S-谷胱甘肽化蛋白质。作为一个重要例子,我们发现ADP-核糖基化因子6(ARF6)在谷胱甘肽耗竭诱导的铁死亡过程中受S-谷胱甘肽化调节。我们的研究结果表明,CHAC1上调降低了ARF6的S-谷胱甘肽化,导致溶酶体中ARF6水平下降。这反过来又增强了转铁蛋白受体(TFRC)在细胞膜上的定位并增加了转铁蛋白摄取,最终损害了ARF6在谷胱甘肽耗竭诱导的铁死亡中的保护作用。使用GalNAc-siTfrc靶向TFRC可减轻体内对乙酰氨基酚诱导的肝损伤。总之,我们的研究提供了证据表明谷胱甘肽池的可用性影响蛋白质S-谷胱甘肽化并调节蛋白质功能以影响铁死亡过程,这为理解谷胱甘肽耗竭诱导的细胞铁死亡开辟了一条途径。