Lai Yimei, Wang Shuang, Ren Tingting, Shi Jia, Qian Yichao, Wang Shuyi, Zhou Mianjing, Watanabe Ryu, Li Mengyuan, Ruan Xinyuan, Wang Xin, Zhuang Lili, Ke Zunfu, Yang Niansheng, Huang Yuefang, Zhang Hui
Department of Rheumatology and Clinical immunology, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China.
Institute of Precision Medicine, the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China.
Nat Commun. 2025 May 15;16(1):4502. doi: 10.1038/s41467-025-59786-z.
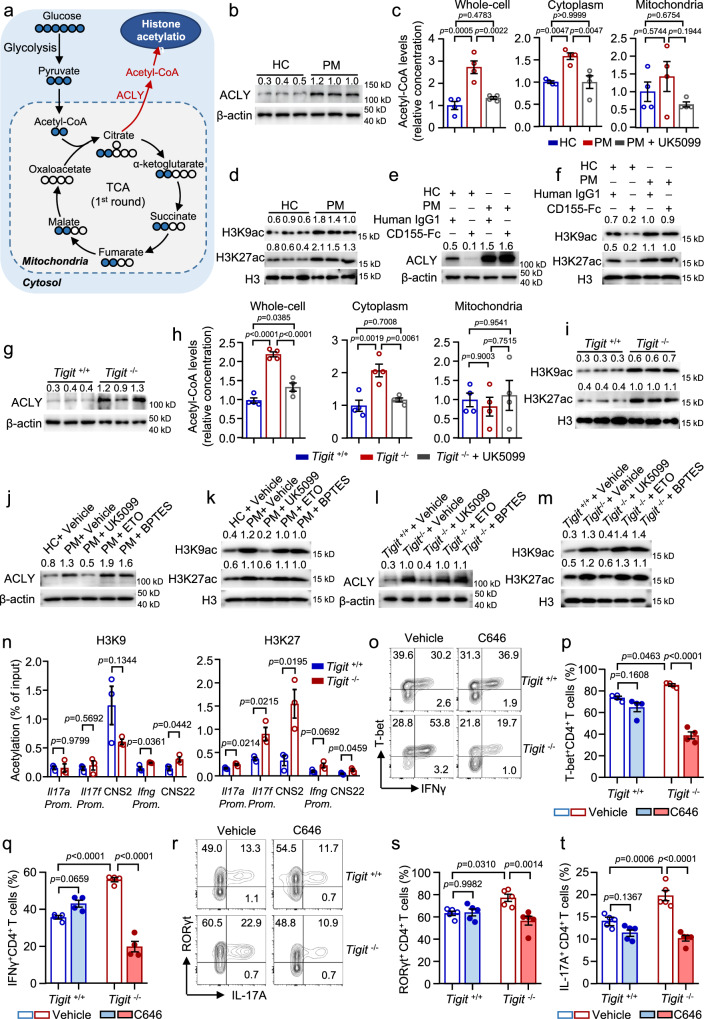
Polymyositis (PM) is a systemic autoimmune disease characterized by muscular inflammatory infiltrates and degeneration. T-cell immunoreceptor with Ig and ITIM domains (TIGIT) contributes to immune tolerance by inhibiting T cell-mediated autoimmunity. Here, we show that a reduced expression of TIGIT in CD4 T cells from patients with PM promotes these cells' differentiation into Th1 and Th17 cells, which could be rescued by TIGIT overexpression. Knockout of TIGIT enhances muscle inflammation in a mouse model of experimental autoimmune myositis. Mechanistically, we find that TIGIT deficiency enhances CD28-mediated PI3K/AKT/mTOR co-stimulatory pathway, which promotes glucose oxidation, citrate production, and increased cytosolic acetyl-CoA levels, ultimately inducing epigenetic reprogramming via histone acetylation. Importantly, pharmacological inhibition of histone acetylation suppresses the differentiation of Th1 and Th17 cells, alleviating muscle inflammation. Thus, our findings reveal a mechanism by which TIGIT directly affects the differentiation of Th1 and Th17 T cells through metabolic‒epigenetic reprogramming, with important implications for treating systemic autoimmune diseases.
多发性肌炎(PM)是一种以肌肉炎症浸润和变性为特征的全身性自身免疫性疾病。具有Ig和ITIM结构域的T细胞免疫受体(TIGIT)通过抑制T细胞介导的自身免疫来促进免疫耐受。在此,我们表明,PM患者CD4 T细胞中TIGIT表达降低会促进这些细胞分化为Th1和Th17细胞,而TIGIT过表达可挽救这一现象。在实验性自身免疫性肌炎小鼠模型中,敲除TIGIT会加剧肌肉炎症。从机制上讲,我们发现TIGIT缺陷会增强CD28介导的PI3K/AKT/mTOR共刺激途径,该途径会促进葡萄糖氧化、柠檬酸盐生成并增加胞质乙酰辅酶A水平,最终通过组蛋白乙酰化诱导表观遗传重编程。重要的是,组蛋白乙酰化的药理学抑制作用可抑制Th1和Th17细胞的分化,减轻肌肉炎症。因此,我们的研究结果揭示了一种机制,即TIGIT通过代谢 - 表观遗传重编程直接影响Th1和Th17 T细胞的分化,这对治疗全身性自身免疫性疾病具有重要意义。