Shu Shangzhi, Fang Junqiao, Zhao Longguo, Han Jiatong, Zhang Meiping, Huang Chaoqun, Cheng Xian Wu, Li Shuyan
Department of Cardiovascular Disease, The First Hospital of Jilin University, Changchun, Jilin, China.
Department of Cardiology and Hypertension, Jilin Provincial Key Laboratory of Stress and Cardiovascular Disease, Yanbian University Hospital, Yanji, Jilin, China.
FASEB J. 2025 May 31;39(10):e70650. doi: 10.1096/fj.202500371R.
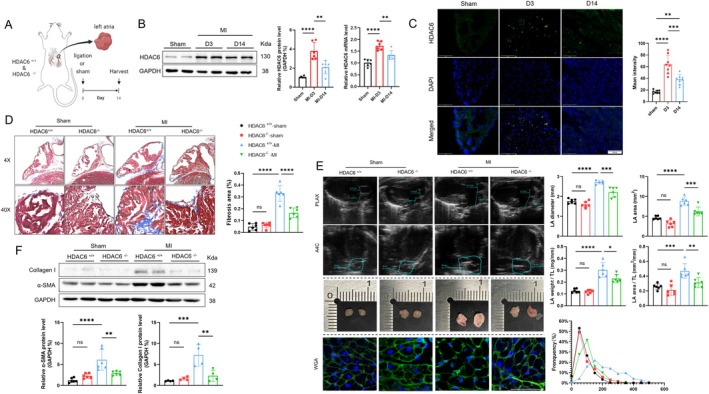
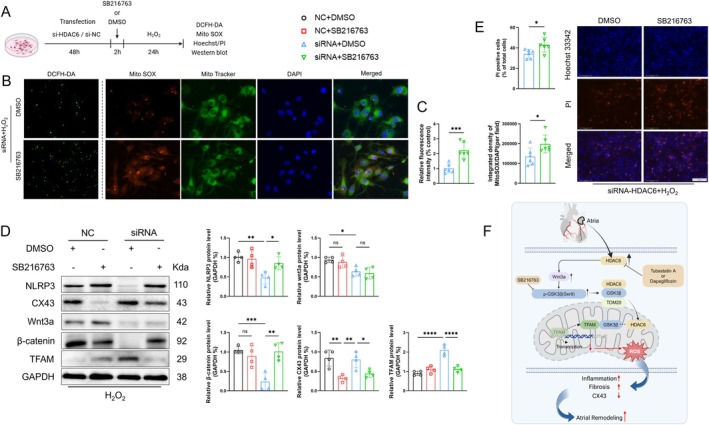
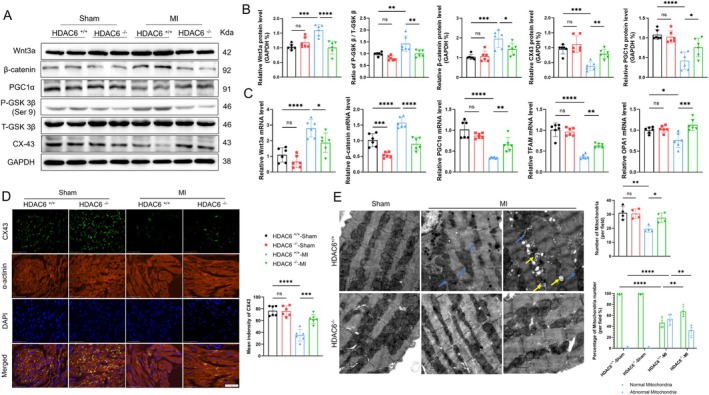
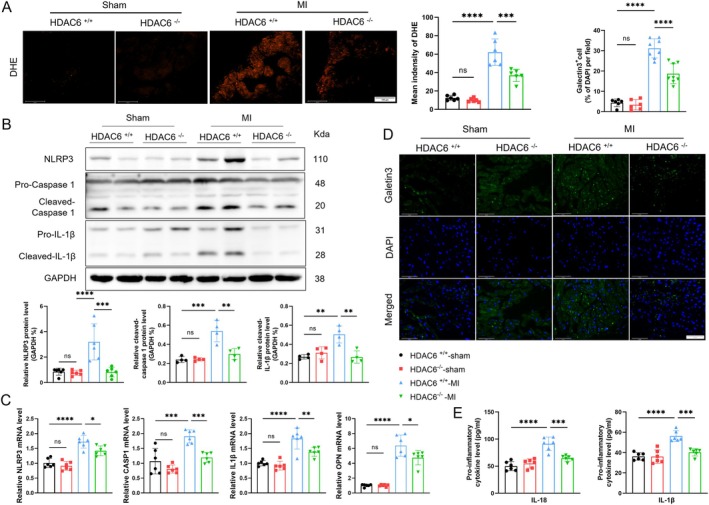
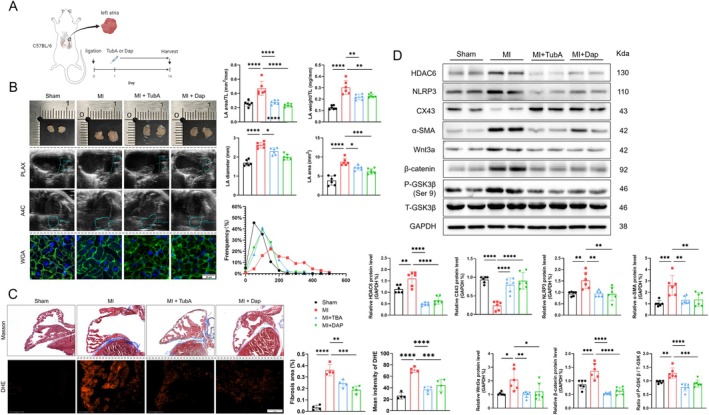
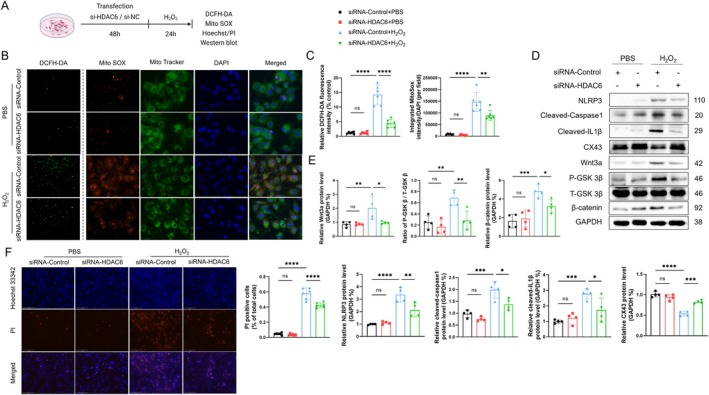
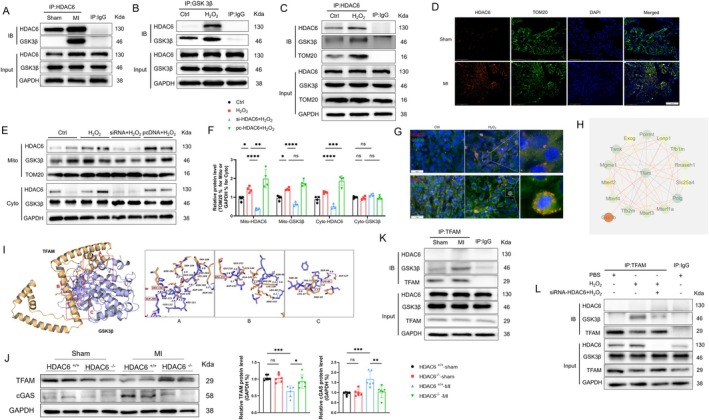
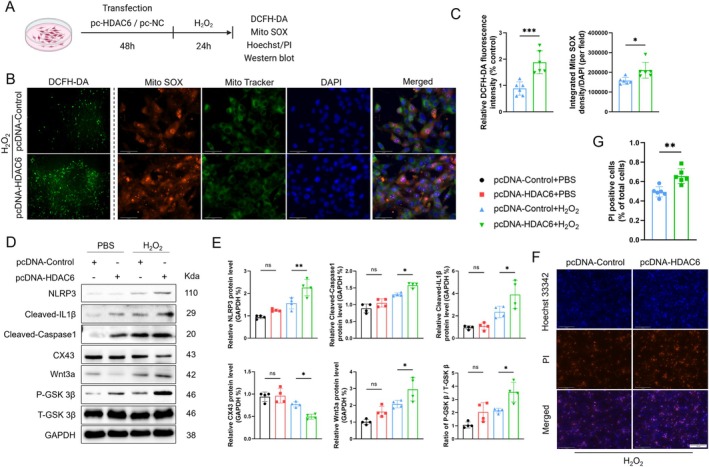
Myocardial infarction (MI)-induced hemodynamic disorder often causes atrial structural and electrophysiological remodeling. Given that histone deacetylase 6 (HDAC6) plays important roles in pathobiology, we investigated the molecular mechanism underlying MI-induced atrial remodeling in mice, with a special focus on HDAC6-mediated Wnt3a/GSK3β signaling activation. We observed an upregulation of HDAC6 expression in the left atria of mice at 2 weeks post-MI, accompanied by atrial enlargement, increased atrial fibrosis and inflammation, myocyte hypertrophy, impaired mitochondrial biogenesis, elevated levels of Wnt3a, GSK3β, and β-catenin protein, and reduced gap junction CX43 expression; these alterations were reversed by HDAC6 deletion. This atrialoprotective effect was mimicked by HDAC6 inhibition with the HDAC6 inhibitor tubastatin A (TubA). In HL1 mouse atrial myocytes, HDAC6 silencing (or overexpression) reduced (increased) the Wnt3a and p-GSK3β protein levels, providing evidence and a mechanistic explanation of HDAC6-mediated Wnt3a/GSK3β signaling activation in mitochondrial oxidative stress production and cell pyroptosis. After HDAC6 formed a complex with GSK3β and translocated into the mitochondria, GSK3β competitively bound with TFAM to mtDNA, thereby affecting mitochondrial function and ROS generation. The SGLT2 inhibitor dapagliflozin exhibited efficacy that was comparable to that of TubA by inhibiting HDAC6 signaling in mice. These results indicate an essential role of HDAC6 in atrial remodeling in response to post-MI stress, possibly via the modulation of Wnt3a/GSK3β-mediated mitochondrial oxidative stress production and pyroptosis and matrix protein production, and they suggest a novel therapeutic strategy for the prevention of post-MI-related atrial morphological and electrophysiological remodeling by regulating HDAC6 activity.
心肌梗死(MI)诱导的血流动力学紊乱常导致心房结构和电生理重构。鉴于组蛋白去乙酰化酶6(HDAC6)在病理生物学中发挥重要作用,我们研究了小鼠MI诱导的心房重构的分子机制,特别关注HDAC6介导的Wnt3a/GSK3β信号激活。我们观察到MI后2周小鼠左心房中HDAC6表达上调,同时伴有心房扩大、心房纤维化和炎症增加、心肌细胞肥大、线粒体生物发生受损、Wnt3a、GSK3β和β-连环蛋白蛋白水平升高以及缝隙连接CX43表达降低;这些改变通过HDAC6缺失得以逆转。HDAC6抑制剂tubastatin A(TubA)抑制HDAC6可模拟这种心房保护作用。在HL1小鼠心房肌细胞中,HDAC6沉默(或过表达)降低(增加)了Wnt3a和p-GSK3β蛋白水平,为HDAC6介导的Wnt3a/GSK3β信号激活在线粒体氧化应激产生和细胞焦亡中的作用提供了证据和机制解释。HDAC6与GSK3β形成复合物并转运到线粒体后,GSK3β与TFAM竞争性结合线粒体DNA,从而影响线粒体功能和活性氧生成。钠-葡萄糖协同转运蛋白2抑制剂达格列净通过抑制小鼠体内的HDAC6信号,表现出与TubA相当的疗效。这些结果表明HDAC6在MI后应激引起的心房重构中起关键作用,可能是通过调节Wnt3a/GSK3β介导的线粒体氧化应激产生、焦亡和基质蛋白生成,并且它们提示了一种通过调节HDAC6活性来预防MI后相关心房形态和电生理重构的新治疗策略。