de Vries Bálint S, de Boer Eva Maria Johanna, Brugman Frans, Van Damme Philip, Veldink Jan Herman, van Es Michael A, van den Berg Leonard H
Department of Neurology, Brain Center Rudolf Magnus, University Medical Center Utrecht, the Netherlands.
ACIBADEM International Medical Center, Amsterdam, the Netherlands.
Neurology. 2025 Jun 10;104(11):e213461. doi: 10.1212/WNL.0000000000213461. Epub 2025 May 19.
Primary lateral sclerosis (PLS) is a rare disease characterized by upper motor neuron (UMN) degeneration. We aimed to elucidate the natural history in patients with UMN syndromes suggestive of PLS and validate the most recent diagnostic (consensus) criteria.
A validation study of a long-term follow-up cohort was conducted, including adults with UMN syndromes and disease durations ≥6 months. Patients were assessed at baseline (T1), at 3 years (T2), and when possible after 13 years (T3). Diagnostic categorization followed the 2020 PLS consensus criteria. Main outcomes included diagnostic classification at follow-up and survival.
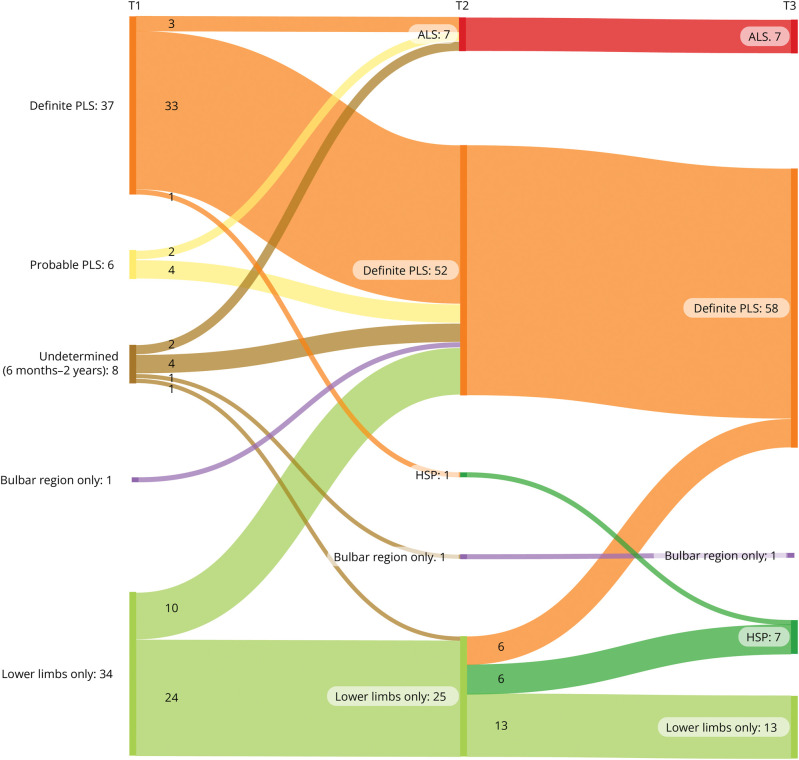
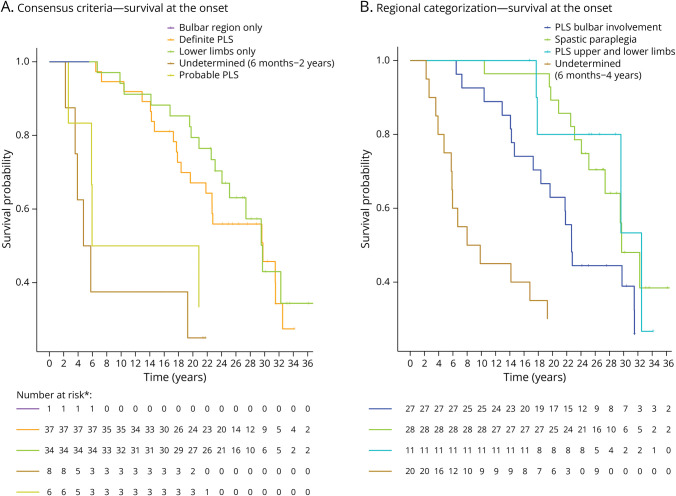
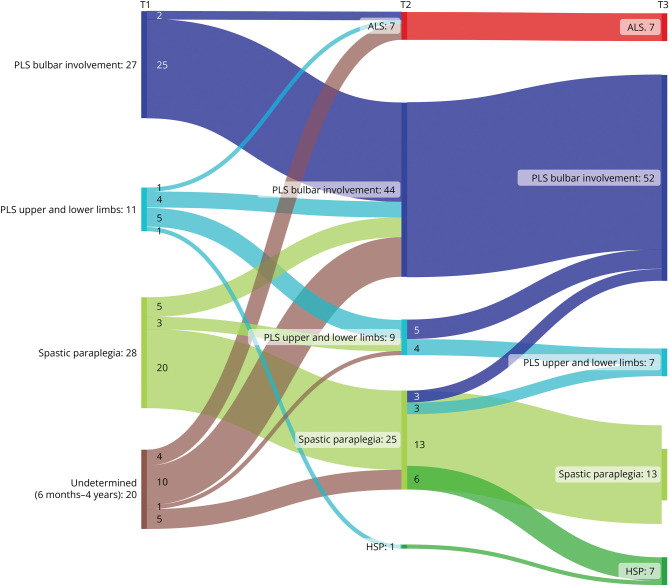
The study comprised 86 patients (34 women [40%], mean age 58.9 ± 10.1 years), of whom 43 met the PLS consensus criteria at baseline (6 probable, 37 definite). Eight patients had a disease duration <2 years, and 35 patients presented with UMN symptoms localized to 1 region (1 bulbar, 34 legs). Change of initial diagnosis occurred in 14% of patients with PLS, and 49% of patients presenting with UMN symptoms in 1 region progressed to PLS. Seven patients developed amyotrophic lateral sclerosis (ALS), and for 7 patients, diagnosis was revised to hereditary spastic paraplegia (HSP). Survival was shorter for patients with a disease duration <4 years. In the probable PLS group, 33% converted to ALS. Converters had a steeper Amyotrophic Lateral Sclerosis Functional Rating Scale slope ( = 0.023) and shorter symptom duration ( < 0.001) at inclusion. Of patients presenting with leg symptoms, diagnosis was revised between T2 and T3 in 29%. Introducing a 4-year minimal disease duration for PLS diagnosis and categorization based on regions involved resulted in 86% of PLS diagnoses remaining within the PLS category, 5% transitioning to ALS (slow variant), and 9% to HSP. Survival was longest for patients presenting with symptoms confined to arms and legs or legs only, followed by those with bulbar involvement at baseline, while patients with disease durations between 6 months and 4 years exhibited the shortest survival.
Our findings suggest that a diagnosis of PLS should be deferred until 4 years after symptom onset because shorter durations correlate with higher ALS conversion rates and shorter survival. Categorization by regional involvement may facilitate more effective monitoring of patients with UMN syndromes.
原发性侧索硬化症(PLS)是一种罕见疾病,以上运动神经元(UMN)变性为特征。我们旨在阐明疑似PLS的UMN综合征患者的自然病史,并验证最新的诊断(共识)标准。
对一个长期随访队列进行了验证研究,纳入患有UMN综合征且病程≥6个月的成年人。在基线期(T1)、3年时(T2)以及尽可能在13年后(T3)对患者进行评估。诊断分类遵循2020年PLS共识标准。主要结局包括随访时的诊断分类和生存率。
该研究包括86例患者(34名女性[40%],平均年龄58.9±10.1岁),其中43例在基线时符合PLS共识标准(6例可能,37例确诊)。8例患者病程<2年,35例患者的UMN症状局限于1个区域(1例延髓,34例腿部)。14%的PLS患者初始诊断发生了变化,1个区域出现UMN症状的患者中有49%进展为PLS。7例患者发展为肌萎缩侧索硬化症(ALS),7例患者的诊断被修订为遗传性痉挛性截瘫(HSP)。病程<4年的患者生存率较短。在可能的PLS组中,33%转变为ALS。转变者在纳入时的肌萎缩侧索硬化症功能评定量表斜率更陡(=0.023)且症状持续时间更短(<0.001)。出现腿部症状的患者中,29%在T2和T3之间诊断被修订。引入4年的最小病程用于基于受累区域的PLS诊断和分类,结果显示86%的PLS诊断仍属于PLS类别,5%转变为ALS(缓慢变异型),9%转变为HSP。症状局限于手臂和腿部或仅腿部的患者生存率最长,其次是基线时有延髓受累的患者,而病程在6个月至4年之间的患者生存率最短。
我们的研究结果表明,PLS的诊断应推迟到症状出现后4年,因为病程较短与较高的ALS转化率和较短的生存率相关。按区域受累进行分类可能有助于更有效地监测UMN综合征患者。