Tedesco Maria Giovanna, Donati Ilaria, Romeo Chiara, Dal Bo Sara, Nardini Chiara, Innoceta Anna Maria, Parmeggiani Giulia, Patanè Anna, Graziano Claudio
Unit of Medical Genetics, Department Maternal-Infantile, S. Maria Della Misericordia Hospital, 06129 Perugia, Italy.
Medical Genetics Unit, MeLabeT Department, AUSL Romagna, 47521 Cesena, Italy.
Genes (Basel). 2025 Apr 29;16(5):524. doi: 10.3390/genes16050524.
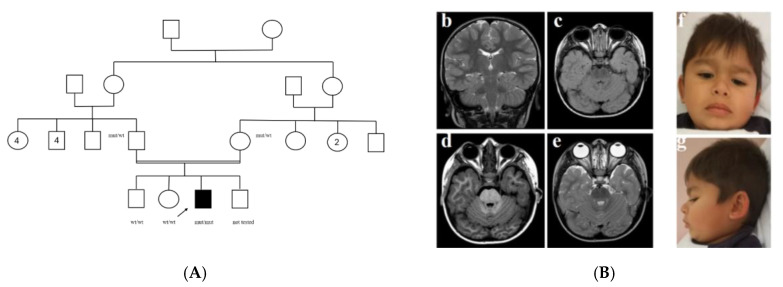
Joubert syndrome (JS) is a multi-systemic ciliopathy, characterized by intellectual disability and congenital anomalies involving the brain, kidney, heart, and eye. Even if clinical presentation is variable, most authors consider a brain abnormality known as the molar tooth sign (MTS) as mandatory for diagnosis. About 40 genes were identified to be associated with JS, usually with an autosomal recessive pattern. variants represent a rare cause of JS; only six families were previously reported. We performed exome sequencing in a child with a syndromic phenotype, described the clinical features and molecular findings, and performed a review of the literature to identify known individuals with pathogenic variants in , highlighting clinical characteristics and gene-phenotype correlations. Using exome sequencing, we identified a homozygous novel frameshift variant c.808del, p.Ser270ValfsTer28 in in a 5-year-old male from a consanguineous family of Roma ethnic background. Notable clinical features of the proband include severe developmental delay, hypotonia, and post-axial polydactyly. He did not have MTS, but showed severe anemia and esophageal atresia, which was already reported in association with a variant. We collected the phenotypes of all reported patients and discussed common and distinct features with respect to typical JS. Affected individuals shared JS clinical features, although the typical MTS was not always present, polydactyly and renal abnormalities were absent, while pituitary abnormalities were common. Our report provides new data for -related JS, expanding the clinical phenotypic spectrum and suggesting a possible role of defects in the development of esophageal atresia.
乔伯特综合征(JS)是一种多系统纤毛病,其特征为智力残疾以及涉及脑、肾、心和眼的先天性异常。即使临床表现存在差异,但大多数作者认为一种称为磨牙征(MTS)的脑异常是诊断的必要条件。已确定约40个基因与JS相关,通常呈常染色体隐性模式。[基因名称]变异是JS的罕见病因;此前仅报道过6个家系。我们对一名具有综合征表型的儿童进行了外显子组测序,描述了临床特征和分子发现,并对文献进行了综述,以识别[基因名称]中具有致病变异的已知个体,突出临床特征和基因 - 表型相关性。通过外显子组测序,我们在一名来自罗姆族近亲家庭的5岁男性中,在[基因名称]中鉴定出一个纯合的新型移码变异c.808del,p.Ser270ValfsTer28。先证者的显著临床特征包括严重发育迟缓、肌张力减退和轴后多指畸形。他没有MTS,但表现出严重贫血和食管闭锁,这在与[基因名称]变异相关的病例中已有报道。我们收集了所有已报道患者的表型,并讨论了与典型JS相关的共同和不同特征。受影响个体具有JS临床特征,尽管并非总是存在典型的MTS,不存在多指畸形和肾脏异常,而垂体异常较为常见。我们的报告为与[基因名称]相关的JS提供了新数据,扩展了临床表型谱,并提示[基因名称]缺陷在食管闭锁发生中可能起的作用。