Genetics and Rare Diseases Research Division, Ospedale Pediatrico Bambino Gesù, IRCCS, Viale di San Paolo 15, 00146, Rome, Italy.
Unit of Endocrinology, Academic Department of Pediatrics, Ospedale Pediatrico Bambino Gesù, Rome, Italy.
BMC Pediatr. 2020 Mar 12;20(1):120. doi: 10.1186/s12887-020-2019-0.
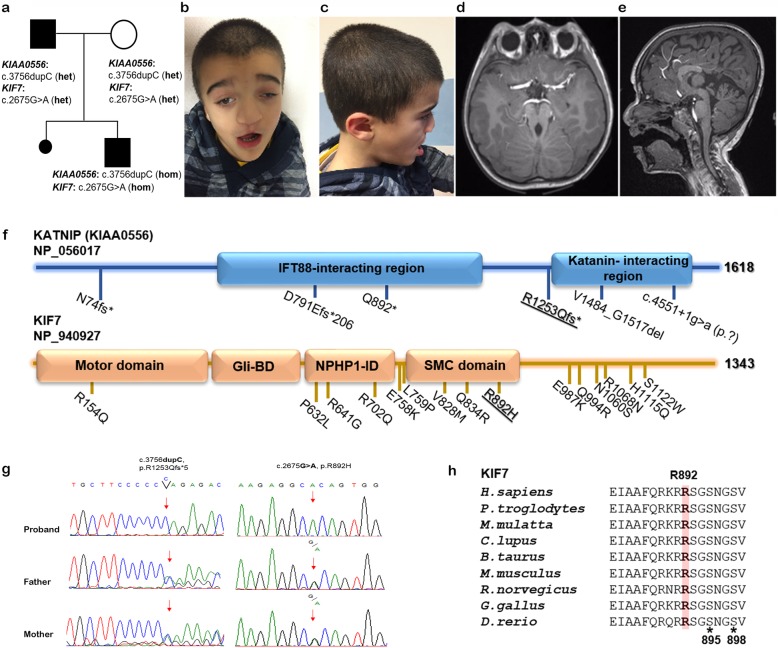
Joubert syndrome is a recessive neurodevelopmental disorder characterized by clinical and genetic heterogeneity. Clinical hallmarks include hypotonia, ataxia, facial dysmorphism, abnormal eye movement, irregular breathing pattern cognitive impairment and, the molar tooth sign is the pathognomonic midbrain-hindbrain malformation on magnetic resonance imaging. The disorder is predominantly caused by biallelic mutations in more than 30 genes encoding proteins with a pivotal role in morphology and function of the primary cilium. Oligogenic inheritance or occurrence of genetic modifiers has been suggested to contribute to the variability of the clinical phenotype. We report on a family with peculiar clinical spectrum Joubert syndrome molecularly and clinically dissecting a complex phenotype, in which hypogonadism, pituitary malformation and growth hormone deficiency occur as major features.
A 7 year-old male was enrolled in a dedicated "Undiagnosed Patients Program" for a peculiar form of Joubert syndrome complicated by iris and retinochoroidal coloboma, hypogonadism pituitary malformation, and growth hormone deficiency. The molecular basis of the complex phenotype was investigated by whole exome sequencing. The concomitant occurrence of homozygosity for mutations in KIF7 and KIAA0556 was identified, and the assessment of major clinical features associated with mutations in these two genes provided evidence that these two independent events represent the cause underlying the complexity of the present clinical phenotype.
Beside the clinical variability of Joubert syndrome, co-occurrence of mutations in ciliopathy-associated genes may contribute to increase the clinical complexity of the trait.
杰伯综合征是一种隐性神经发育障碍,具有临床和遗传异质性。临床特征包括肌张力低下、共济失调、面型异常、眼球运动异常、呼吸节律不规则、认知障碍和磨牙征,磁共振成像上的特征性中脑-后脑畸形。该疾病主要由编码原发性纤毛蛋白的 30 多个基因的双等位基因突变引起,这些蛋白在原发性纤毛的形态和功能中起着关键作用。寡基因遗传或遗传修饰物的发生被认为有助于临床表型的可变性。我们报告了一个家族,该家族具有独特的临床谱杰伯综合征分子上和临床上剖析了一种复杂的表型,其中性腺功能减退、垂体畸形和生长激素缺乏是主要特征。
一名 7 岁男性因一种特殊形式的杰伯综合征(伴有虹膜和视网膜脉络膜裂孔)、性腺功能减退、垂体畸形和生长激素缺乏而被纳入专门的“未确诊患者计划”。通过全外显子组测序研究了复杂表型的分子基础。确定了 KIF7 和 KIAA0556 基因突变的纯合性,对这些两种基因突变相关的主要临床特征的评估提供了证据,表明这两个独立事件是导致目前临床表型复杂性的原因。
除了杰伯综合征的临床变异性外,纤毛病相关基因的突变共存可能有助于增加该特征的临床复杂性。