Mochales-Riaño Gabriel, Hirst Samuel R, Talavera Adrián, Burriel-Carranza Bernat, Pagone Viviana, Estarellas Maria, Busschau Theo, Boissinot Stéphane, Hogan Michael P, Tena-Garcés Jordi, Pla Davinia, Calvete Juan J, Els Johannes, Margres Mark J, Carranza Salvador
Institute of Evolutionary Biology (IBE), CSIC-Universitat Pompeu Fabra, 08003 Barcelona, Spain.
Department of Integrative Biology, University of South Florida, Tampa, FL 33620, USA.
Gigascience. 2025 Jan 6;14. doi: 10.1093/gigascience/giaf030.
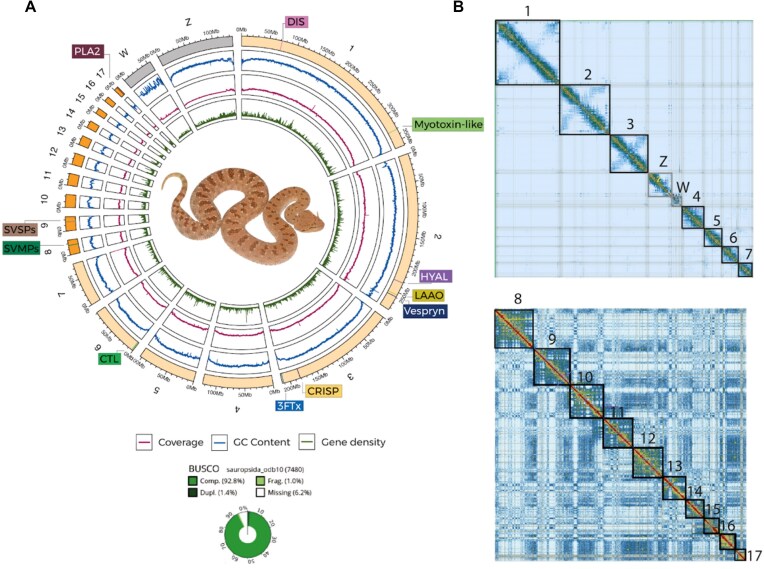
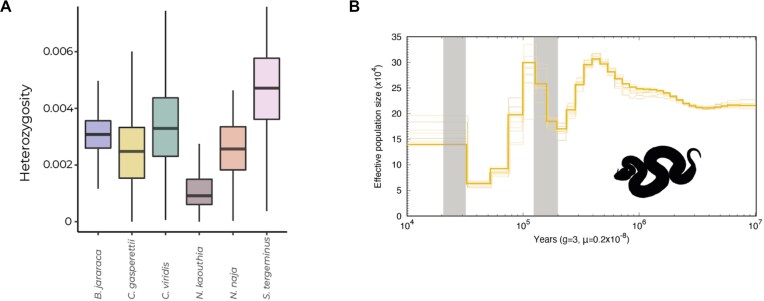
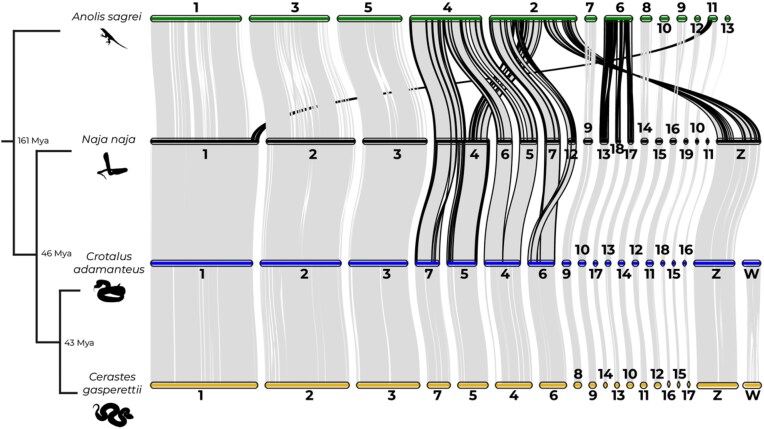
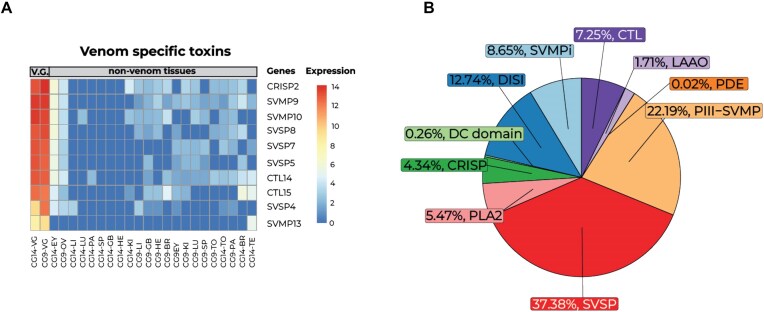
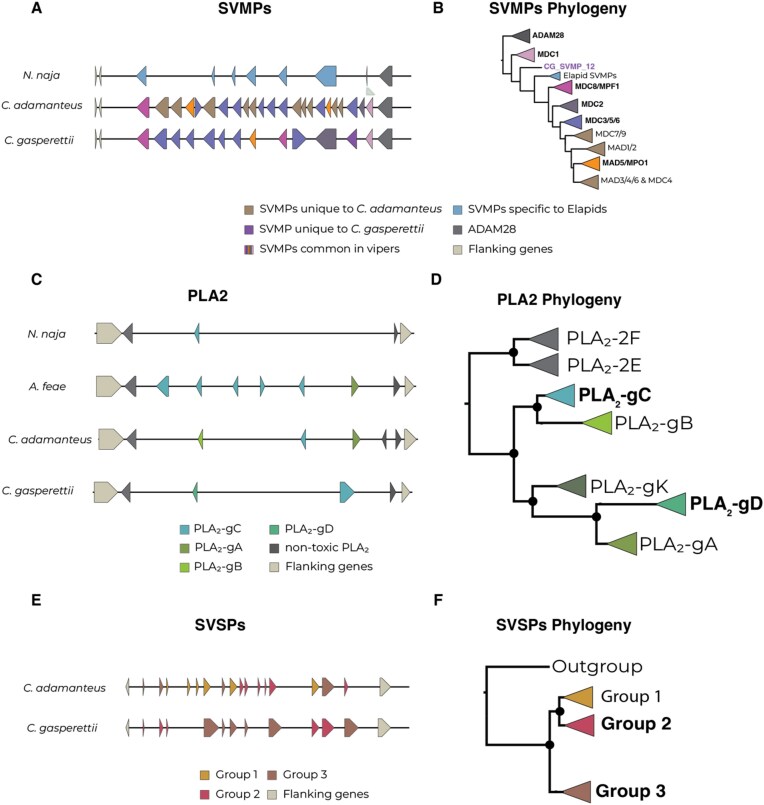
Venoms have traditionally been studied from a proteomic and/or transcriptomic perspective, often overlooking the true genetic complexity underlying venom production. The recent surge in genome-based venom research (sometimes called "venomics") has proven to be instrumental in deepening our understanding of venom evolution at the molecular level, particularly through the identification and mapping of toxin-coding loci across the broader chromosomal architecture. Although venomous snakes are a model system in venom research, the number of high-quality reference genomes in the group remains limited. In this study, we present a chromosome-resolution reference genome for the Arabian horned viper Cerastes gasperettii (NCBI: txid110202), a venomous snake native to the Arabian Peninsula. Our highly contiguous genome (genome size: 1.63 Gbp; contig N50: 45.6 Mbp; BUSCO: 92.8%) allowed us to explore macrochromosomal rearrangements within the Viperidae family, as well as across squamates. We identified the main highly expressed toxin genes within the venom glands comprising the venom's core, in line with our proteomic results. We also compared microsyntenic changes in the main toxin gene clusters with those of other venomous snake species, highlighting the pivotal role of gene duplication and loss in the emergence and diversification of snake venom metalloproteinases and snake venom serine proteases for C. gasperettii. Using Illumina short-read sequencing data, we reconstructed the demographic history and genome-wide heterozigosity of the species, revealing how historical aridity likely drove population expansions. Finally, this study highlights the importance of using long-read sequencing as well as chromosome-level reference genomes to disentangle the origin and diversification of toxin gene families in venomous snake species.
传统上,人们从蛋白质组学和/或转录组学的角度研究毒液,常常忽略了毒液产生背后真正的遗传复杂性。最近基于基因组的毒液研究(有时称为“毒液组学”)激增,已被证明有助于加深我们在分子水平上对毒液进化的理解,特别是通过在更广泛的染色体结构中识别和定位毒素编码基因座。尽管毒蛇是毒液研究中的一个模型系统,但该类群中高质量参考基因组的数量仍然有限。在本研究中,我们展示了阿拉伯角蝰Cerastes gasperettii(NCBI:txid110202)的染色体水平参考基因组,这是一种原产于阿拉伯半岛的毒蛇。我们高度连续的基因组(基因组大小:1.63 Gbp;重叠群N50:45.6 Mbp;BUSCO:92.8%)使我们能够探索蝰蛇科内部以及有鳞目动物之间的宏观染色体重排。我们确定了毒液腺内主要的高表达毒素基因,这些基因构成了毒液的核心,这与我们的蛋白质组学结果一致。我们还比较了主要毒素基因簇与其他毒蛇物种的微同线性变化,突出了基因复制和丢失在加氏角蝰蛇毒金属蛋白酶和蛇毒丝氨酸蛋白酶的出现和多样化中的关键作用。利用Illumina短读长测序数据,我们重建了该物种的种群历史和全基因组杂合度,揭示了历史干旱可能如何推动种群扩张。最后,本研究强调了使用长读长测序以及染色体水平参考基因组来解开毒蛇物种中毒素基因家族的起源和多样化的重要性。