Avilés A, Beltran S Perez, Ghotbi M, Ferguson A J, Blackburn J L, Darensbourg M Y, Balbuena P B
Chemistry and Nanoscience Center, National Renewable Energy Laboratory, Golden, Colorado 80401, United States.
J Phys Chem Lett. 2025 Jun 19;16(24):6125-6137. doi: 10.1021/acs.jpclett.5c00812. Epub 2025 Jun 10.
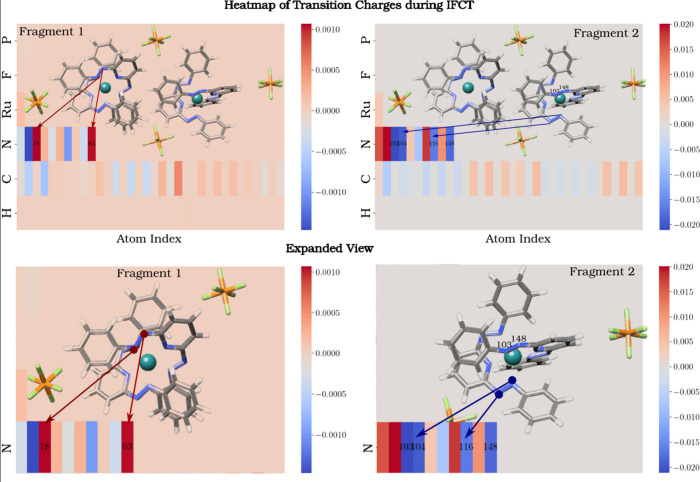
Redox-driven conductance changes are critical processes in molecular- and coordination-complex-based memristive thin films and devices that are envisioned for neuromorphic technologies, but fundamental mechanisms of conductance switching are not fully understood. Here, we explore charge disproportionation (CD) processes in RuL molecular systems that intrinsically involve interfragment charge transfer (IFCT). Using a combination of molecular dynamics simulation (AIMD), time-dependent density functional theory (TD-DFT), and density functional theory (DFT) calculations, we investigate the electron transfer mechanisms and the roles of temperature and cell volumetric expansion in facilitating the counterion movements and electronic transitions required for low-cost IFCT and charge redistribution. A detailed analysis of the density of states and TD-DFT calculations highlights that unpaired electrons play a crucial role in low-energy transitions, with the azo (N═N) groups of the ligand serving as the primary sites for electronic transport between molecular fragments, further stabilizing the asymmetric state. Localization of added electrons on azo ligands occurs with negligible change at the Ru centers, supported by atomic volume expansions up to +4.74 bohr, and goes along with a progressive reduction of the HOMO-LUMO gap across redox states, suggesting enhanced conductivity. The TD-DFT analysis reveals a dominant IFCT excitation at 2082.76 nm in the doubly reduced (22) state, while a stabilization energy of 1.20 eV of the asymmetric (13) state relative to the symmetric (22) state is predicted by constrained DFT. Periodic DFT and AIMD simulations emulating a molecular film show that the stabilization of the asymmetric state, relative to a symmetric one, translates in net charge separation values (order of ∼0.33 e) that are strongly linked to increased counterion mobility (average counterion displacements exceeding 0.7 Å per atom during CD events) and the involvement of azo groups in electron redistribution. These findings, which align with previously reported experimental and computational data, provide key insights into the IFCT mechanisms and electronic transport facilitated by azo groups, with important implications for redox-driven memristive and neuromorphic technologies.
氧化还原驱动的电导变化是基于分子和配位络合物的忆阻薄膜及器件中的关键过程,这些薄膜和器件被设想用于神经形态技术,但电导切换的基本机制尚未完全理解。在这里,我们探索了RuL分子体系中的电荷歧化(CD)过程,该过程本质上涉及片段间电荷转移(IFCT)。通过结合分子动力学模拟(AIMD)、含时密度泛函理论(TD-DFT)和密度泛函理论(DFT)计算,我们研究了电子转移机制以及温度和晶胞体积膨胀在促进低成本IFCT和电荷重新分布所需的抗衡离子移动和电子跃迁中的作用。对态密度的详细分析和TD-DFT计算表明,未成对电子在低能跃迁中起关键作用,配体的偶氮(N═N)基团是分子片段之间电子传输的主要位点,进一步稳定了不对称状态。添加的电子在偶氮配体上的定位在Ru中心处变化可忽略不计,原子体积膨胀高达+4.74玻尔支持了这一点,并且随着氧化还原状态下HOMO-LUMO能隙的逐渐减小,表明导电性增强。TD-DFT分析揭示了在双还原(22)状态下2082.76 nm处的主导IFCT激发,而通过受限DFT预测不对称(13)状态相对于对称(22)状态的稳定能为1.20 eV。模拟分子薄膜的周期性DFT和AIMD模拟表明,相对于对称状态,不对称状态的稳定转化为净电荷分离值(约为0.33 e量级),这与抗衡离子迁移率增加(CD事件期间每个原子的平均抗衡离子位移超过0.7 Å)以及偶氮基团参与电子重新分布密切相关。这些发现与先前报道实验和计算数据一致,为IFCT机制以及偶氮基团促进的电子传输提供了关键见解,对氧化还原驱动的忆阻和神经形态技术具有重要意义。