Shivakumar Vikram S, Langmead Ben
Department of Computer Science, Johns Hopkins University, Baltimore, USA.
Genome Biol. 2025 Jun 17;26(1):169. doi: 10.1186/s13059-025-03644-0.
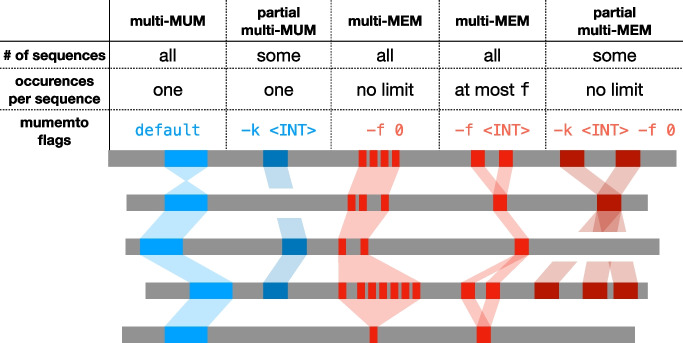
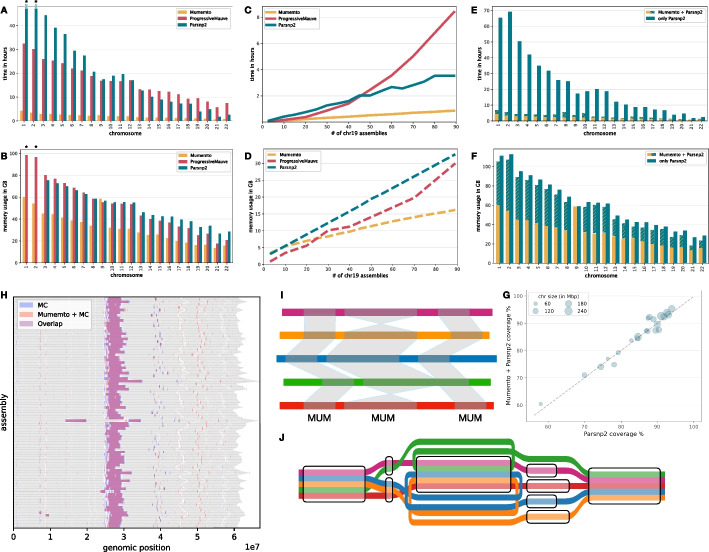
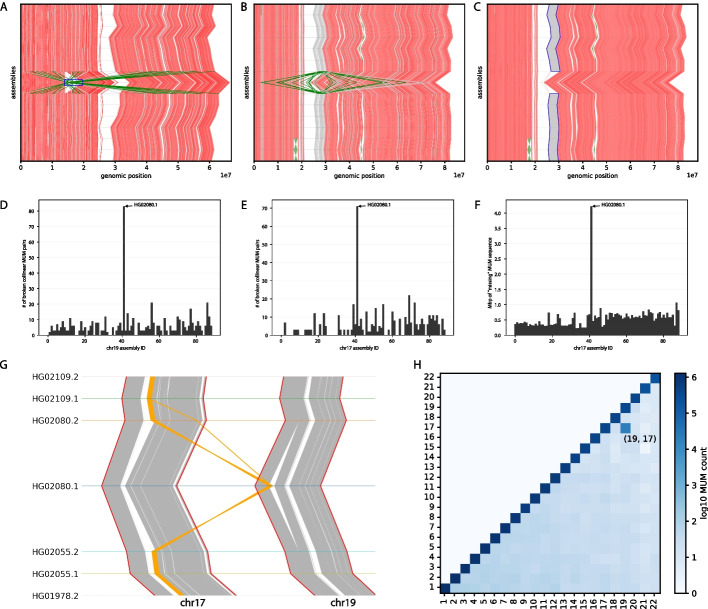
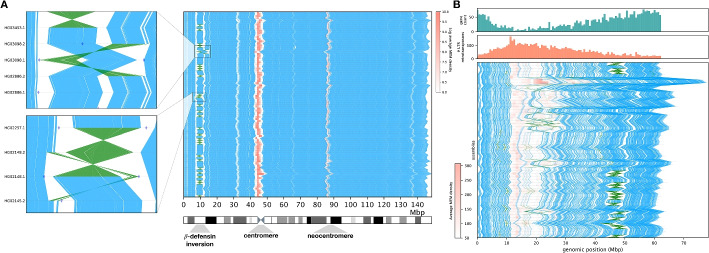
Aligning genomes into common coordinates is central to pangenome construction, though computationally expensive. Multi-sequence maximal unique matches (multi-MUMs) help to frame and solve the multiple alignment problem. We introduce Mumemto, a tool that computes multi-MUMs and other match types across large pangenomes. Mumemto allows for visualization of synteny, reveals aberrant assemblies and scaffolds, and highlights pangenome conservation and structural variation. Mumemto computes multi-MUMs across 320 human assemblies (960GB) in 25.7 h with 800 GB of memory and hundreds of fungal assemblies in minutes. Mumemto is implemented in C++ and Python and available open-source at https://github.com/vikshiv/mumemto (v1.1.1 at doi.org/10.5281/zenodo.15053447 ).
将基因组比对到共同的坐标是构建泛基因组的核心,尽管计算成本高昂。多序列最大唯一匹配(multi-MUMs)有助于构建和解决多重比对问题。我们引入了Mumemto,这是一种可在大型泛基因组中计算多MUMs和其他匹配类型的工具。Mumemto允许对共线性进行可视化,揭示异常组装和支架,并突出显示泛基因组的保守性和结构变异。Mumemto使用800GB内存,在25.7小时内可对320个人类基因组组装(960GB)计算多MUMs,对数百个真菌基因组组装只需几分钟。Mumemto用C++和Python实现,可在https://github.com/vikshiv/mumemto(doi.org/10.5281/zenodo.15053447上的v1.1.1版本)开源获取。