Aleksandra Sobuń, Marta Madej, Ewa Mroczek, Maciej Guziński, Piotr Wiland, Agata Sebastian
Department of Rheumatology and Internal Medicine, University Clinical Hospital in Wroclaw, Wroclaw, Poland.
Department of Rheumatology and Internal Medicine, Wroclaw Medical University, Wroclaw, Poland.
Rheumatol Int. 2025 Jun 26;45(7):159. doi: 10.1007/s00296-025-05896-2.
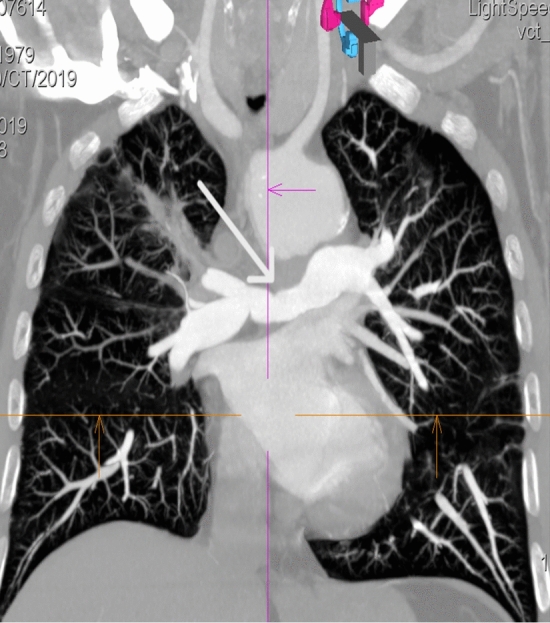
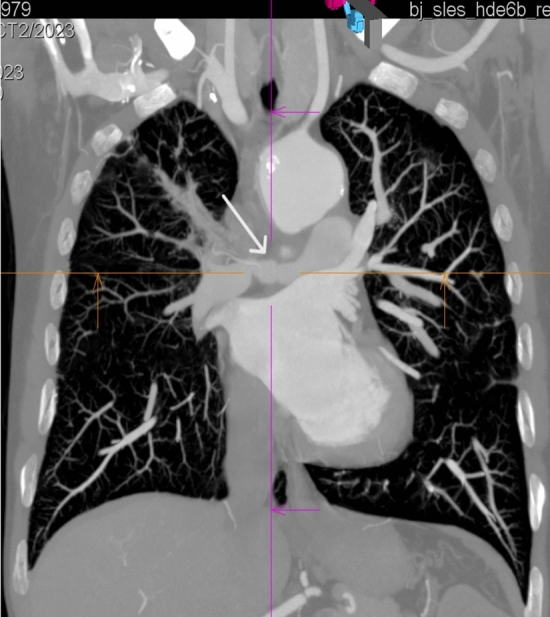
Granulomatosis with polyangiitis (GPA) is a rare disease that belongs to the group of necrotizing systemic vasculitis, which is characterized by the involvement of small and medium-sized blood vessels, the formation of granulomas, and the presence of proteinase 3-anti-neutrophil cytoplasmic antibodies (PR3-ANCA). The disease typically involves the upper and lower respiratory tract and kidneys, but other organs and systems may also be involved. A rare manifestation of the disease is the involvement of large vessels, which may occur in the form of aneurysms, dissection and stenosis. We present the case of a 39-year-old male patient who was diagnosed with GPA several years ago, with the involvement of lungs, kidneys, skin and inflammation of the ascending aorta. The patient currently reported fever, cough and dyspnea. The imaging studies showed stenosis of the right and left pulmonary arteries. The treatment included glucocorticosteroids and rituximab. A review of the literature on pulmonary artery involvement in granulomatosis with polyangiitis was performed, seven case reports meeting such criteria were found.
肉芽肿性多血管炎(GPA)是一种罕见疾病,属于坏死性系统性血管炎,其特征为中小血管受累、肉芽肿形成以及蛋白酶3抗中性粒细胞胞浆抗体(PR3-ANCA)阳性。该疾病通常累及上、下呼吸道及肾脏,但其他器官和系统也可能受累。该疾病的一种罕见表现是大血管受累,可能以动脉瘤、夹层和狭窄的形式出现。我们报告一例39岁男性患者,数年前被诊断为GPA,累及肺、肾、皮肤及升主动脉炎症。患者目前诉发热、咳嗽和呼吸困难。影像学检查显示左右肺动脉狭窄。治疗包括糖皮质激素和利妥昔单抗。我们对关于肉芽肿性多血管炎累及肺动脉的文献进行了回顾,发现了7篇符合此类标准的病例报告。