Hamilton Erika P, Patel Manish R, Borges Virginia F, Meisel Jane L, Okera Meena, Alemany Carlos A, Pluard Timothy J, Wesolowski Robert, Sabanathan Dhanusha, Miller Kathy D, Conlin Alison K, McCarthy Nicole, Shaw Morena, Tonda Margaret, Shilkrut Mark, Lin Nancy U
Sarah Cannon Research Institute, 335 24th Ave North Ste 300, Nashville, TN, 37203, USA.
Florida Cancer Specialists/Sarah Cannon Research Institute, Sarasota, FL, USA.
Breast Cancer Res. 2025 Jul 1;27(1):119. doi: 10.1186/s13058-025-02049-y.
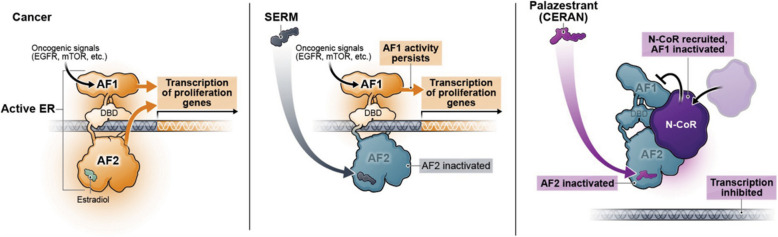
Endocrine resistance is a major challenge in treating patients with ER+ /HER2- metastatic breast cancer (MBC) necessitating a switch from endocrine therapy to more toxic therapies. Mutations in ESR1 constitute a key mechanism of resistance to endocrine therapy in ER+ /HER2- BC. Therapies that overcome endocrine resistance are needed. Palazestrant is a novel oral complete estrogen receptor (ER) antagonist (CERAN) and selective ER degrader (SERD) belonging to a new class of ER-targeting agents that completely blocks estrogen-induced transcriptional activity, regardless of ESR1 mutation status. This first-in-human, open-label, multicenter, phase 1/2 dose-escalation/expansion study was designed to determine the recommended phase 2 dose (RP2D) and to evaluate safety, pharmacokinetics, and antitumor activity of palazestrant in patients with ER+ /HER2- MBC with disease progression on prior treatment.
Adults with ER+ /HER2‒ MBC who received ≥ 1 prior line of endocrine therapy for advanced disease and ≤ 2 prior chemotherapy regimens for metastatic disease were eligible. Patients received once-daily oral palazestrant (30-300 mg) in 28-day cycles until progression or intolerable toxicity.
This study enrolled 146 patients. No dose-limiting toxicities were observed at doses up to 300 mg/day palazestrant. Confirmed partial responses were observed with 60 and 120 mg/day palazestrant. Both doses showed similar and tolerable safety profiles, favorable pharmacokinetics, and steady-state plasma concentrations above the predicted threshold for complete ER inhibition. Greater clinical benefit at palazestrant 120 mg/day (46%) versus 60 mg/day (19%) led to selection of 120 mg/day as RP2D and study expansion dose. At 120 mg/day, the median progression-free survival was 4.8 months (95% CI, 3.5-7.1) overall and 5.6 months (95% CI, 4.8-NE) among patients with cancers with ESR1 mutations. Most treatment-emergent adverse events (TEAEs) were grade 1-2. The most common TEAEs were nausea (62.8%), vomiting (29.1%), and fatigue (25.6%). The most common grade ≥ 3 TEAE was transient neutropenia (10.5%) managed by dose interruption and reduction.
Palazestrant demonstrated a manageable safety profile, with antitumor activity observed in patients with heavily pretreated cancers with wild-type and ESR1-mutated BC. These data support the ongoing phase 3 study evaluating palazestrant in patients with ER+ /HER2 - MBC.
ClinicalTrials.gov, NCT04505826 . Registered August 6, 2020.
内分泌抵抗是治疗雌激素受体阳性(ER+)/人表皮生长因子受体2阴性(HER2-)转移性乳腺癌(MBC)患者的一项重大挑战,这使得治疗需要从内分泌治疗转向毒性更强的治疗方法。ESR1基因突变是ER+ /HER2- 乳腺癌内分泌治疗耐药的关键机制。因此,需要能够克服内分泌抵抗的治疗方法。帕拉泽斯特兰是一种新型口服全雌激素受体(ER)拮抗剂(CERAN)和选择性ER降解剂(SERD),属于一类新型的ER靶向药物,无论ESR1突变状态如何,均可完全阻断雌激素诱导的转录活性。这项首次人体、开放标签、多中心、1/2期剂量递增/扩展研究旨在确定推荐的2期剂量(RP2D),并评估帕拉泽斯特兰在先前治疗后疾病进展的ER+ /HER2- MBC患者中的安全性、药代动力学和抗肿瘤活性。
符合条件的患者为患有ER+ /HER2- MBC的成年人,这些患者接受过≥1线晚期疾病内分泌治疗,且接受过≤2线转移性疾病化疗方案。患者在28天周期内每日口服一次帕拉泽斯特兰(30-300 mg),直至疾病进展或出现无法耐受的毒性。
本研究共纳入146例患者。在帕拉泽斯特兰剂量高达300 mg/天时,未观察到剂量限制性毒性。在帕拉泽斯特兰剂量为60和120 mg/天时观察到确认的部分缓解。两种剂量均显示出相似且可耐受的安全性、良好的药代动力学,以及高于完全抑制ER预测阈值的稳态血浆浓度。帕拉泽斯特兰剂量为120 mg/天时的临床获益率(46%)高于60 mg/天时(19%),因此选择120 mg/天作为RP2D和研究扩展剂量。在120 mg/天时,总体无进展生存期(PFS)中位数为4.8个月(95%CI,3.5-7.1),ESR1基因突变的癌症患者中PFS中位数为5.6个月(95%CI,4.8-未达到)。大多数治疗期间出现的不良事件(TEAE)为1-2级。最常见的TEAE为恶心(62.8%)、呕吐(29.1%)和疲劳((25.6%)。最常见的≥3级TEAE为短暂性中性粒细胞减少(10.5%),通过剂量中断和减少进行处理。
帕拉泽斯特兰显示出可控的安全性,在野生型和ESR1突变型乳腺癌的重度预处理患者中观察到抗肿瘤活性。这些数据支持正在进行的评估帕拉泽斯特兰治疗ER+ /HER2- MBC患者的3期研究。
ClinicalTrials.gov,NCT04505826。2020年8月6日注册。