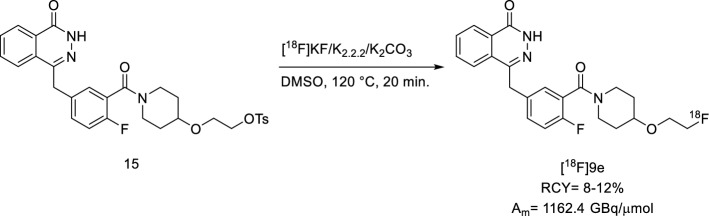
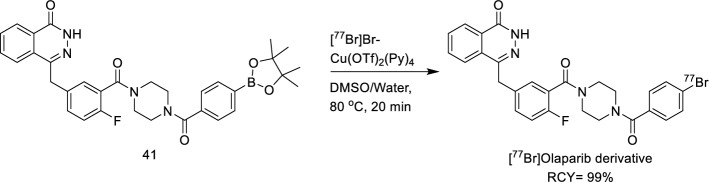
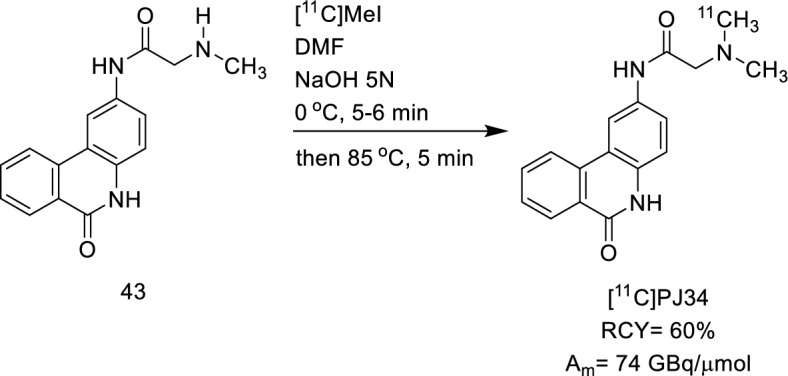
Destro Gianluca, Rizzo Rebecca, Rua Chiara, Azimi Raha Rouhbakhsh, Morbelli Silvia
Department of Molecular Biotechnology and Health Sciences, University of Turin, Piazza Nizza 44/Bis, 10126, Turin, Italy.
Department of Nuclear Medicine, University of Turin, Turin, Italy.
EJNMMI Radiopharm Chem. 2025 Jul 3;10(1):37. doi: 10.1186/s41181-025-00364-5.
Poly (ADP-ribose) polymerase (PARP) inhibitors have emerged as a promising class of therapeutics, particularly in the treatment of cancers with defective DNA repair mechanisms, such as those with breast cancer genes (BRCA) mutations. Their effectiveness in cancer therapy is now well-established, but the ongoing advancements in radiochemistry are expanding their potential to combine both therapeutic and imaging capabilities. Radiolabelled PARP inhibitors, used in conjunction with positron emission tomography (PET) or single-photon emission computed tomography (SPECT), might enable precise imaging of PARP expression in tumours, potentially providing invaluable insights into treatment response, tumor heterogeneity, and molecular profiling.
The radiochemistry of PARP inhibitors involves incorporating radioisotopes (most of all Fluorine-18) into the molecular structure of these molecules. The first strategy used to achieve this goal was the use of prosthetic groups bearing the fluorine-18. Then, the development of radioisotopologue have gained ground, followed later by the replacement with other halogens such as bromine, iodine, or astatine has taken place. Another frontier is represented by the metal radiolabelling of these inhibitors through the introduction of a chelator moiety to these molecules, thus further expanding both imaging and therapy applications.
Finally, emerging evidence suggest the possibility to involve PARP-related radiopharmaceuticals in theranostics approaches. Despite challenges such as the complexity of radiolabelling, regulatory hurdles, and the need for more robust clinical validation, the continued exploration of the radiochemistry of PARP inhibitors promises to revolutionize both the diagnosis and treatment of cancer, offering hope for more effective and personalized cancer care.
聚(ADP - 核糖)聚合酶(PARP)抑制剂已成为一类很有前景的治疗药物,尤其在治疗具有缺陷DNA修复机制的癌症方面,例如那些具有乳腺癌基因(BRCA)突变的癌症。它们在癌症治疗中的有效性现已得到充分证实,但放射化学的不断进步正在扩大其结合治疗和成像能力的潜力。与正电子发射断层扫描(PET)或单光子发射计算机断层扫描(SPECT)结合使用的放射性标记PARP抑制剂,可能能够精确成像肿瘤中PARP的表达, potentially提供有关治疗反应、肿瘤异质性和分子谱的宝贵见解。
PARP抑制剂的放射化学涉及将放射性同位素(最重要的是氟 - 18)纳入这些分子的分子结构中。实现这一目标的第一种策略是使用带有氟 - 18的辅基。然后,放射性同位素类似物的开发逐渐兴起,随后又发生了用其他卤素如溴、碘或砹进行取代。另一个前沿领域是通过向这些抑制剂引入螯合剂部分来对其进行金属放射性标记,从而进一步扩大成像和治疗应用。
最后,新出现的证据表明PARP相关放射性药物有可能用于诊疗方法。尽管存在诸如放射性标记的复杂性、监管障碍以及需要更有力的临床验证等挑战,但对PARP抑制剂放射化学的持续探索有望彻底改变癌症的诊断和治疗,为更有效和个性化的癌症护理带来希望。