Chen Ziheng, Liu Yaxuan, Brown Ashley R, Sestili Heather H, Ramamurthy Easwaran, Xiong Xushen, Prokopenko Dmitry, Phan BaDoi N, Gadey Lahari, Hu Peinan, Tsai Li-Huei, Bertram Lars, Hide Winston, Tanzi Rudolph E, Kellis Manolis, Pfenning Andreas R
bioRxiv. 2025 Jul 24:2025.07.11.659973. doi: 10.1101/2025.07.11.659973.
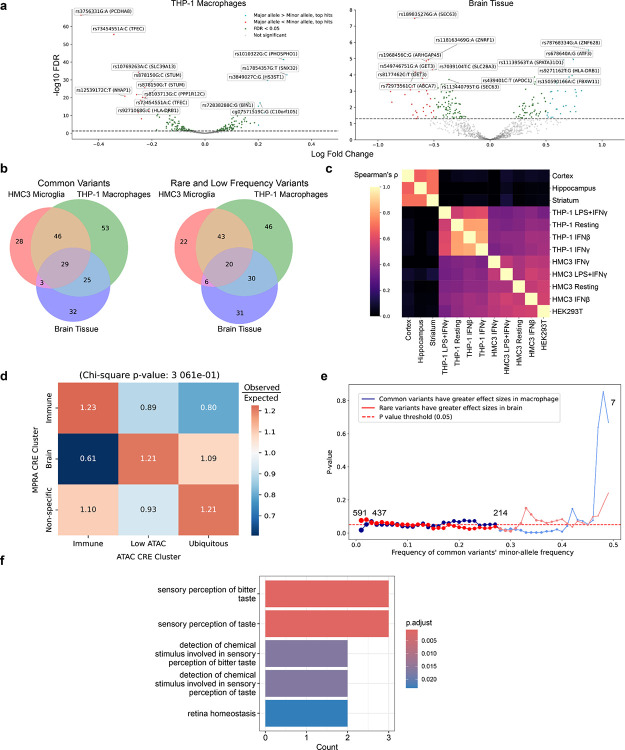
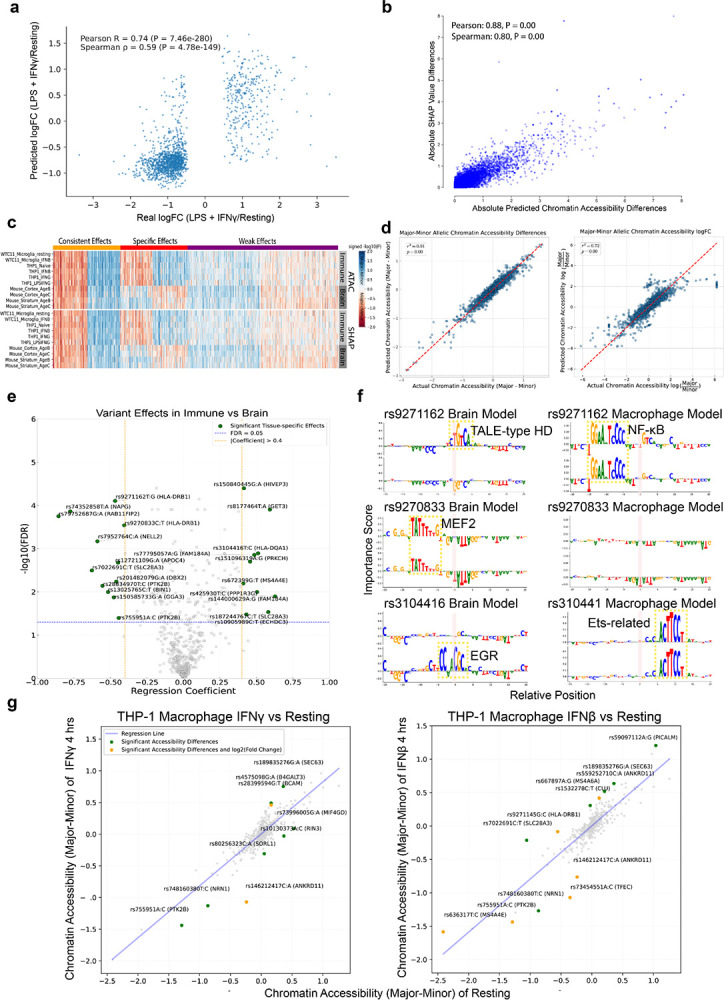
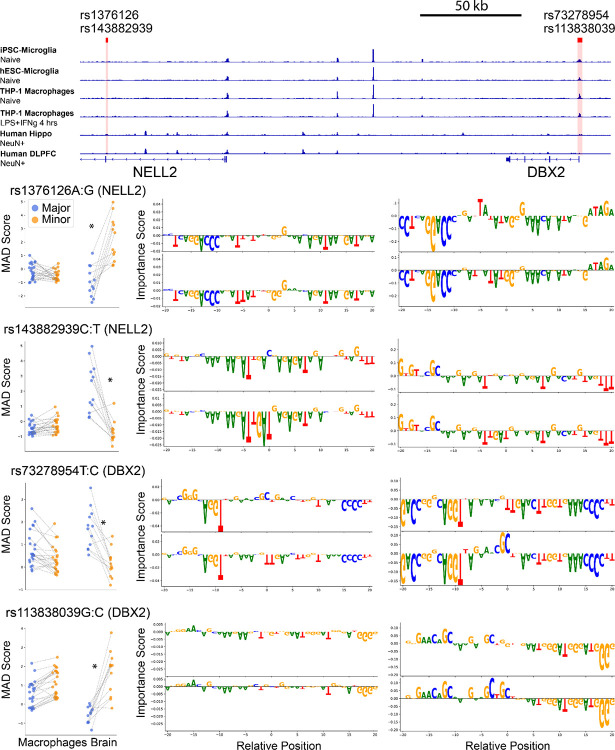
Noncoding genetic variants underlie many complex diseases, yet identifying and interpreting their functional impacts remains challenging. Late-onset Alzheimer's disease (LOAD), a polygenic neurodegenerative disorder, exemplifies this challenge. The disease is strongly associated with noncoding variation, including common variants enriched in microglial enhancers and rare variants that are hypothesized to influence neurodevelopment and synaptic plasticity. These variants often perturb regulatory sequences by disrupting transcription factor (TF) motifs or altering local TF interactions, thereby reshaping gene expression and chromatin accessibility. However, assessing their impact is complicated by the context-dependent functions of regulatory sequences, underscoring the need to systematically examine variant effects across diverse tissues, cell types, and cellular states. Here, we combined and massively parallel reporter assays (MPRAs) with interpretable machine-learning models to systematically characterize common and rare variants across myeloid and neural contexts. Parallel profiling of variants in four immune states and three mouse brain regions revealed that individual variants can differentially and even oppositely modulate regulatory function depending on cell-type and cell-state contexts. Common variants associated with LOAD tended to exert stronger effects in immune contexts, whereas rare variants showed more pronounced impacts in brain contexts. Interpretable sequence-to-function deep-learning models elucidated how genetic variation leads to cell-type-specific differences in regulatory activity, pinpointing both direct transcription-factor motif disruptions and subtler tuning of motif context. To probe the broader functional consequences of a locus prioritized by our reporter assays and models, we used CRISPR interference to silence an enhancer within the locus that harbors four functional rare variants, revealing its gatekeeper role in inflammation and amyloidogenesis. These findings underscore the context-dependent nature of noncoding variant effects in LOAD and provide a generalizable framework for the mechanistic interpretation of risk alleles in complex diseases.
非编码基因变异是许多复杂疾病的基础,但识别和解释它们的功能影响仍然具有挑战性。晚发性阿尔茨海默病(LOAD)是一种多基因神经退行性疾病,就是这一挑战的典型例子。该疾病与非编码变异密切相关,包括在小胶质细胞增强子中富集的常见变异以及据推测会影响神经发育和突触可塑性的罕见变异。这些变异通常通过破坏转录因子(TF)基序或改变局部TF相互作用来扰乱调控序列,从而重塑基因表达和染色质可及性。然而,调控序列的上下文依赖性功能使得评估它们的影响变得复杂,这突出了系统检查不同组织、细胞类型和细胞状态下变异效应的必要性。在这里,我们将[未提及具体内容]和大规模平行报告基因检测(MPRAs)与可解释的机器学习模型相结合,以系统地表征髓系和神经环境中的常见和罕见变异。对四种免疫状态[未提及具体内容]和三个小鼠脑区[未提及具体内容]中的变异进行平行分析发现,单个变异根据细胞类型和细胞状态背景可以有差异地甚至相反地调节调控功能。与LOAD相关的常见变异在免疫环境中往往发挥更强的作用,而罕见变异在脑环境中表现出更明显的影响。可解释的序列到功能深度学习模型阐明了基因变异如何导致调控活性的细胞类型特异性差异,确定了直接的转录因子基序破坏和基序上下文的更细微调节。为了探究我们的报告基因检测和模型确定的一个位点的更广泛功能后果,我们使用CRISPR干扰来沉默位于[未提及具体内容]位点内的一个增强子,该增强子含有四个功能性罕见变异,揭示了其在炎症和淀粉样蛋白生成中的守门人作用。这些发现强调了LOAD中非编码变异效应的上下文依赖性,并为复杂疾病中风险等位基因的机制解释提供了一个可推广的框架。