Radhakrishnan Shiva T, Mullish Benjamin H, Olbei Marton L, Danckert Nathan P, Valdivia-Garcia Maria A, Serrano-Contreras Jose I, Chrysostomou Despoina, Balarajah Sharmili, Perry Robert W, Thomas John P, Potari-Gul Lejla, Modos Dezso, Hicks Lucy C, Powell Nick, Orchard Timothy R, Li Jia V, Marchesi Julian R, Korcsmaros Tamas, Alexander James L, Williams Horace R T
Division of Digestive Diseases, Department of Metabolism, Digestion and Reproduction, Faculty of Medicine, Imperial College London, London, UK.
Departments of Gastroenterology and Hepatology, St Mary's Hospital, Imperial College Healthcare NHS Trust, London, UK.
Gut Microbes. 2025 Dec;17(1):2527863. doi: 10.1080/19490976.2025.2527863. Epub 2025 Jul 18.
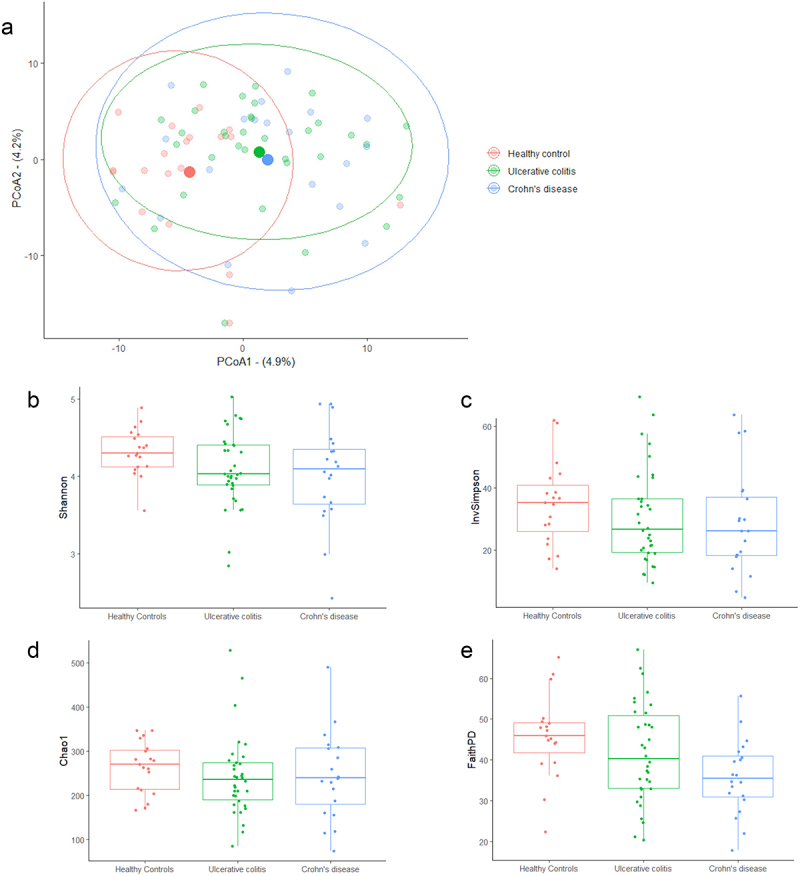
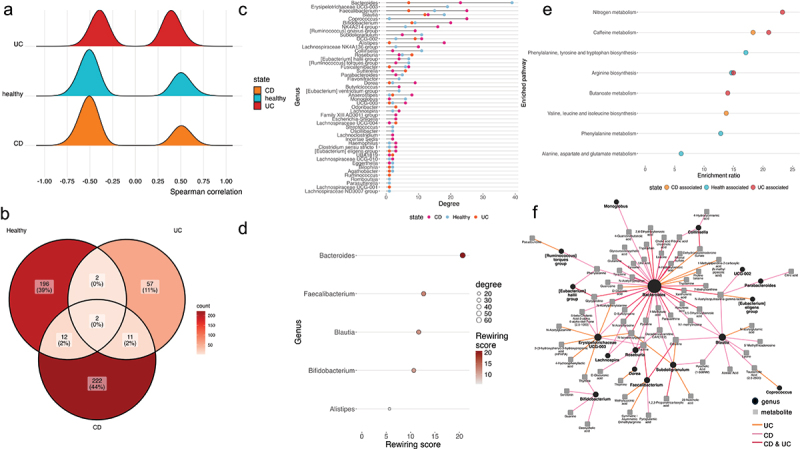
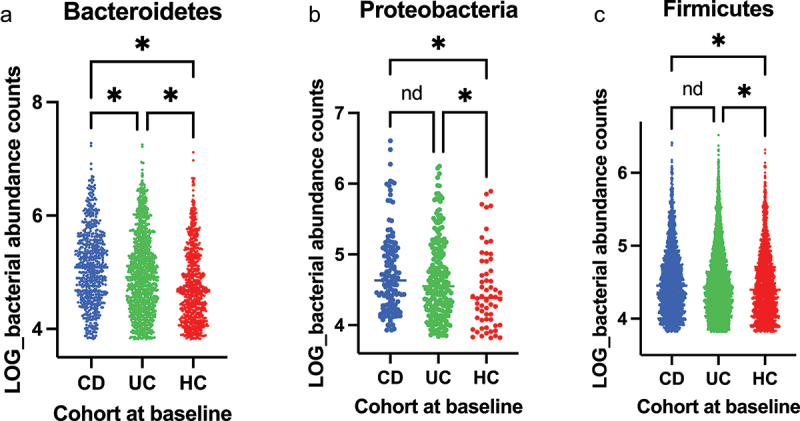
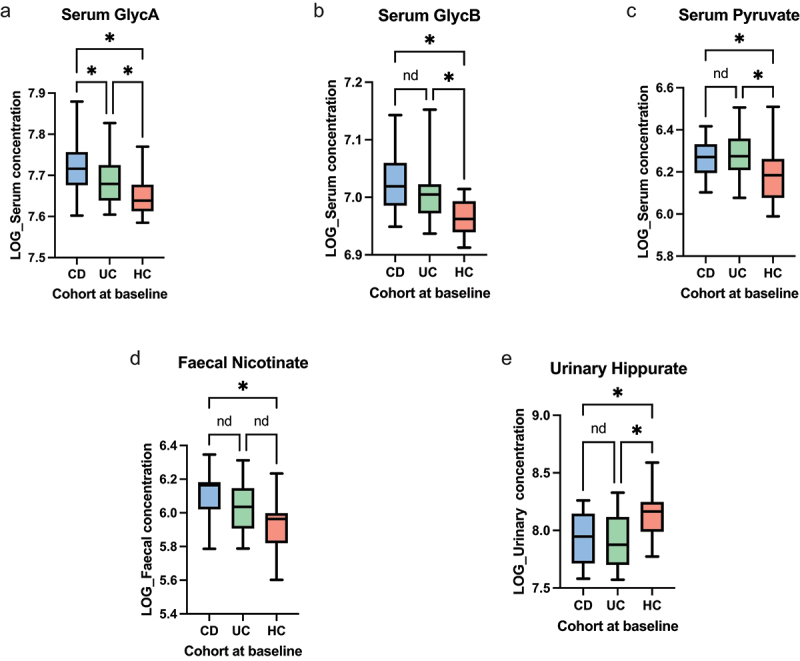
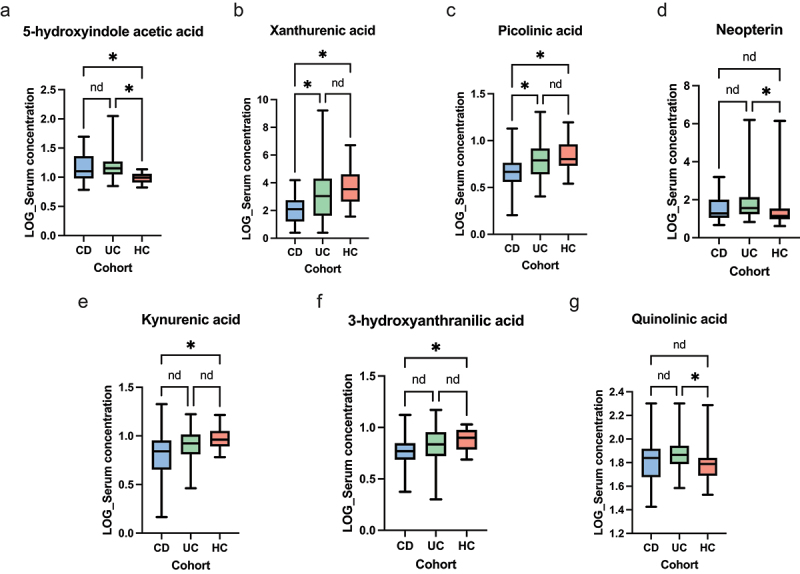
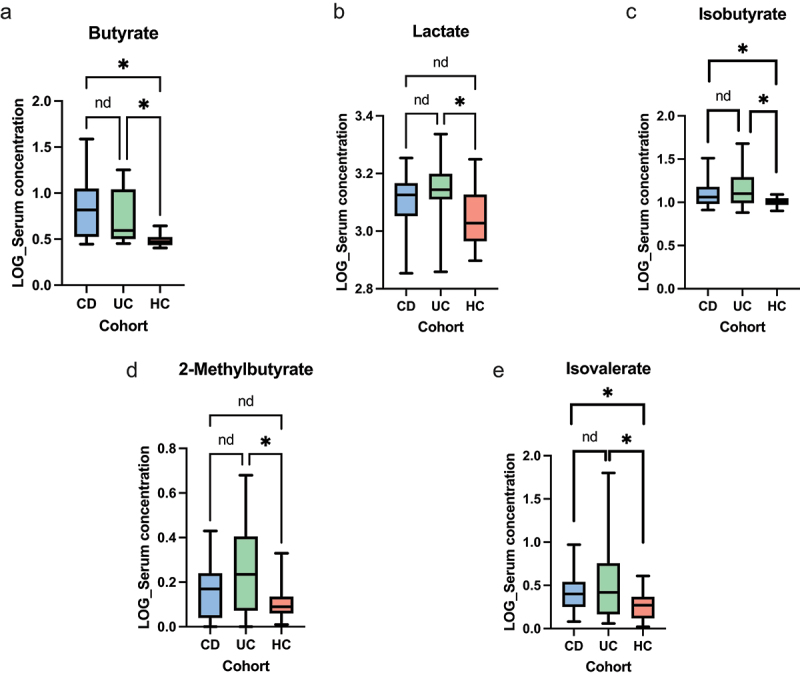
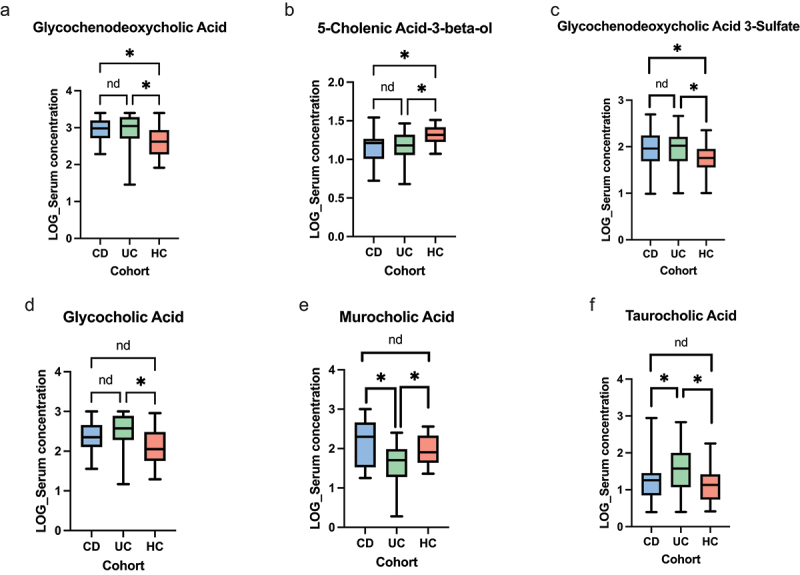
The gut microbiota contribute to the etiopathogenesis of inflammatory bowel disease (IBD), but limitations of prior studies include the use of sequencing alone (restricting exploration of the contribution of microbiota functionality) and the recruitment of patients with well-established disease (introducing potential confounders, such as immunomodulatory medication). Here, we analyze a true IBD inception cohort and healthy controls (HCs) via stool 16S rRNA gene sequencing and multi-system metabolomic phenotyping (using nuclear magnetic spectroscopy and mass spectroscopy), with subsequent integrative network analysis employed to delineate novel microbiota-metabolome interactions in IBD. Marked differences in β diversity and taxonomic profiles were observed both between IBD and HCs, as well as between Crohn's disease (CD) and ulcerative colitis (UC) patients. Multiple between-group metabolomic differences were also observed, particularly with regard to tryptophan-/indole-related metabolites; for example, UC patients had higher levels of serum metabolites including xanthurenic acid ( = 0.0092) and picolinic acid ( = 0.018). Network analysis demonstrated multiple unique interactions in CD compared to HCs with minimal overlap, indicating a loss of 'health-associated' interactions in CD. Compared to HCs, UC patients demonstrated increased pathway activity related to nitrogen and butanoate metabolism, whilst CD patients displayed increased leucine and valine synthesis. Networks from IBD patients overall showed negative correlation with health-specific associations, including an increase in taurine metabolism. Collectively, this work characterizes multiple novel perturbed microbiota-metabolome interactions that are present even at the diagnosis of IBD, which may inform potential future targets to aid diagnosis and direct therapeutic options.
肠道微生物群在炎症性肠病(IBD)的发病机制中发挥作用,但既往研究存在局限性,包括仅使用测序方法(限制了对微生物群功能贡献的探索)以及招募已确诊疾病的患者(引入了潜在的混杂因素,如免疫调节药物)。在此,我们通过粪便16S rRNA基因测序和多系统代谢组学表型分析(使用核磁共振光谱和质谱)对一个真正的IBD发病队列和健康对照(HCs)进行分析,随后采用综合网络分析来描绘IBD中新型的微生物群-代谢组相互作用。在IBD患者与HCs之间以及克罗恩病(CD)和溃疡性结肠炎(UC)患者之间均观察到β多样性和分类学特征存在显著差异。还观察到多组间的代谢组差异,特别是在色氨酸/吲哚相关代谢物方面;例如,UC患者血清代谢物水平较高,包括黄尿酸(P = 0.0092)和吡啶甲酸(P = 0.018)。网络分析表明,与HCs相比,CD中有多种独特的相互作用,重叠极少,表明CD中“与健康相关”的相互作用丧失。与HCs相比,UC患者显示出与氮和丁酸代谢相关的途径活性增加,而CD患者则表现出亮氨酸和缬氨酸合成增加。IBD患者的网络总体上与健康特异性关联呈负相关,包括牛磺酸代谢增加。总的来说,这项研究描绘了即使在IBD诊断时就存在的多种新型的微生物群-代谢组相互作用紊乱,这可能为未来潜在的诊断靶点和指导治疗选择提供依据。