Yang Ji, Guo Peng, Luo Hongtao, Tang Xin, Liu Wei, Ren Xiaolin
College of Laboratory Medicine, Chengdu Medical College, Chengdu, China.
Department of Clinical Medicine, Chengdu Medical College, Chengdu, China.
Front Pharmacol. 2025 Jul 11;16:1614978. doi: 10.3389/fphar.2025.1614978. eCollection 2025.
Current therapeutic options for inflammatory bowel disease (IBD) remain suboptimal due to limited efficacy, significant side effects, and high relapse rates, necessitating novel treatment strategies. Lumefantrine, a clinically established antimalarial drug, emerges as a compelling repurposing candidate based on its putative anti-inflammatory activity, though its efficacy and mechanism in IBD remain unexplored.
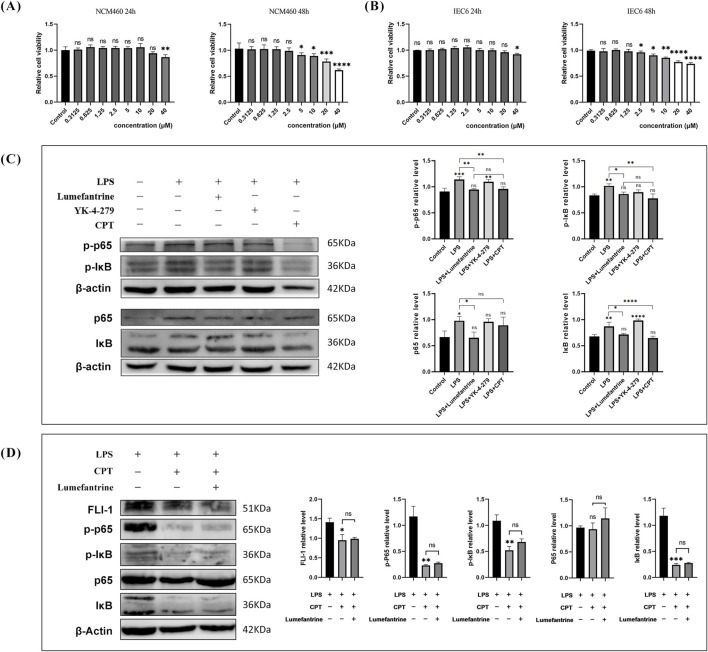
A murine IBD model was induced by 3% dextran sulfate sodium (DSS). Mice received oral Lumefantrine (20 mg/kg/day) for 7 days. Disease progression was monitored via disease activity index (DAI) scoring and histological analysis. Serum cytokines (IL-1β, IL-6, TNF-α) and colonic inflammatory mediators (Cox-2, iNos) were quantified by ELISA and qPCR. Tight junction proteins (Claudin-1, ZO-1) were assessed by immunohistochemistry and Western blot. Molecular targets were identified through computational docking and pull-down assays. Additionally, NF-κB signaling modulation was assessed in lipopolysaccharide (LPS)-stimulated intestinal epithelial cells (IEC-6 and NCM460) via Western blot analysis.
Oral administration of Lumefantrine significantly attenuated disease activity index (DAI) scores and restored intestinal barrier integrity through upregulation of epithelial tight junction proteins Claudin-1 and ZO-1. Treated mice exhibited reduced serum levels of IL-1β, IL-6 and TNF-α, along with decreased colonic expression of inflammatory mediators cyclooxygenase-2 (Cox-2) and inducible nitric oxide synthase (iNos). Computational and experimental approaches identified FLI-1 a transcription factor upregulated in IBD colon tissues as Lumefantrine's direct binding target. This interaction mediated suppression of NF-κB signaling, specifically downregulating phosphorylation of IκBα and p65 in LPS-stimulated intestinal epithelial cells.
Lumefantrine ameliorates experimental colitis through FLI-1-dependent inhibition of the NF-κB pathway, demonstrating high repurposing potential as an IBD therapeutic.
由于疗效有限、副作用显著以及复发率高,炎症性肠病(IBD)目前的治疗选择仍不尽人意,因此需要新的治疗策略。氯喹,一种临床已证实的抗疟药物,基于其假定的抗炎活性,成为一种引人注目的重新利用的候选药物,尽管其在IBD中的疗效和机制仍未得到探索。
用3%硫酸葡聚糖钠(DSS)诱导小鼠IBD模型。小鼠口服氯喹(20毫克/千克/天),持续7天。通过疾病活动指数(DAI)评分和组织学分析监测疾病进展。通过酶联免疫吸附测定(ELISA)和定量聚合酶链反应(qPCR)对血清细胞因子(白细胞介素-1β、白细胞介素-6、肿瘤坏死因子-α)和结肠炎症介质(环氧化酶-2、诱导型一氧化氮合酶)进行定量分析。通过免疫组织化学和蛋白质免疫印迹法评估紧密连接蛋白(闭合蛋白-1、闭锁小带蛋白-1)。通过计算机对接和下拉试验鉴定分子靶点。此外,通过蛋白质免疫印迹分析评估脂多糖(LPS)刺激的肠上皮细胞(IEC-6和NCM460)中核因子κB(NF-κB)信号通路的调节。
口服氯喹显著降低疾病活动指数(DAI)评分,并通过上调上皮紧密连接蛋白闭合蛋白-1和闭锁小带蛋白-1恢复肠道屏障完整性。治疗后的小鼠血清白细胞介素-1β、白细胞介素-6和肿瘤坏死因子-α水平降低,同时结肠炎症介质环氧化酶-2(Cox-2)和诱导型一氧化氮合酶(iNos)的表达减少。计算机和实验方法确定了IBD结肠组织中上调的转录因子Fli-1为氯喹的直接结合靶点。这种相互作用介导了NF-κB信号通路的抑制,特别是下调了LPS刺激的肠上皮细胞中IκBα和p65的磷酸化。
氯喹通过Fli-1依赖的NF-κB途径抑制作用改善实验性结肠炎,显示出作为IBD治疗药物的高重新利用潜力。