Sendrowski Janek, Bataillon Thomas, Ramstein Guillaume P
Bioinformatics Research Center, Aarhus University, 8000, Aarhus, Denmark.
Center for Quantitative Genetics and Genomics, Aarhus University, 8000, Aarhus, Denmark.
Theor Appl Genet. 2025 Jul 28;138(8):193. doi: 10.1007/s00122-025-04973-1.
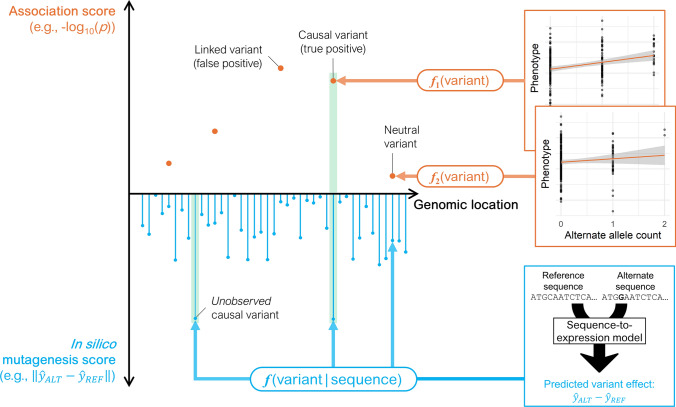
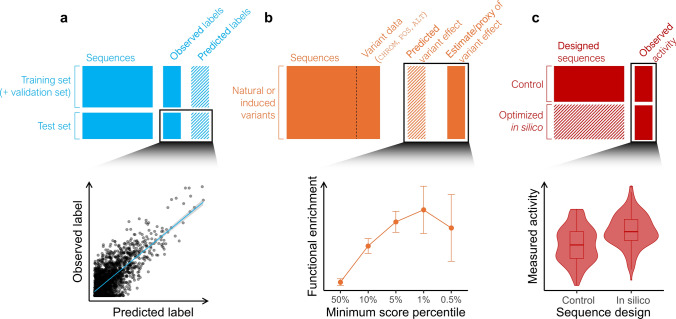
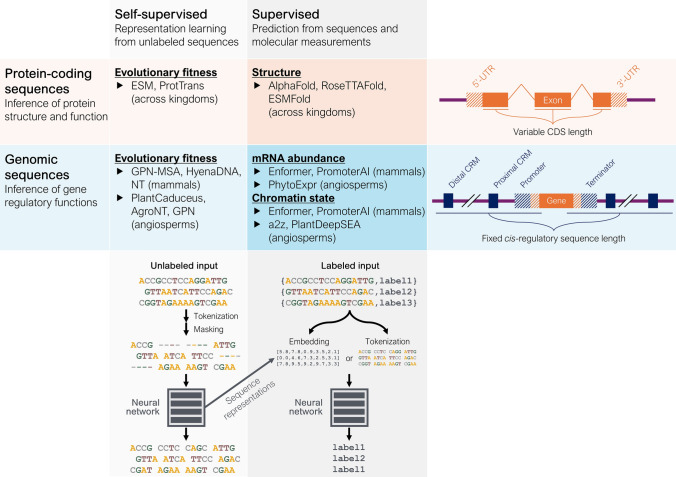
Sequence-based AI models show great potential for prediction of variant effects at high resolution, but their practical value in plant breeding remains to be confirmed through rigorous validation studies. Plant breeding has traditionally relied on phenotyping to select individuals with desirable traits-a process that is both costly and time-consuming. Increasingly, breeding strategies are shifting toward precision breeding, where causal variants are directly targeted based on their effects. To predict the effects of causal variants, in silico methods are emerging as efficient alternatives or complements to mutagenesis screens. Here, we review state-of-the-art machine learning methods for predicting variant effects in plants across both coding and noncoding regions, contrasting supervised approaches in functional genomics with unsupervised methods in comparative genomics. We discuss challenges in validating predictions, and compare these methods with traditional association and comparative genomics techniques. We argue that modern sequence models extend traditional methods by generalizing across genomic contexts, fitting a unified model across loci rather than a separate model for each locus. In doing so, they address inherent limitations of traditional quantitative and evolutionary comparative genetics techniques. However, the accuracy and generalizability of sequence models heavily depend on the training data, highlighting the need for validation experiments. We point to successful applications of sequence models, especially with protein sequences, and identify areas for further improvement, especially in modeling regulatory sequences. While not yet mature for in silico-driven precision breeding, sequence models show strong potential to become an integral part of the breeder's toolbox.
基于序列的人工智能模型在高分辨率预测变异效应方面显示出巨大潜力,但其在植物育种中的实际价值仍有待通过严格的验证研究来证实。传统上,植物育种依赖于表型分析来选择具有理想性状的个体,这一过程既昂贵又耗时。越来越多的育种策略正朝着精准育种转变,即根据因果变异的效应直接靶向这些变异。为了预测因果变异的效应,计算机模拟方法正成为诱变筛选的有效替代方法或补充方法。在这里,我们综述了用于预测植物编码区和非编码区变异效应的最新机器学习方法,对比了功能基因组学中的监督方法和比较基因组学中的无监督方法。我们讨论了验证预测结果时面临的挑战,并将这些方法与传统的关联分析和比较基因组学技术进行了比较。我们认为,现代序列模型通过在基因组背景下进行泛化,扩展了传统方法,为各个位点拟合一个统一的模型,而不是为每个位点分别拟合一个模型。通过这样做,它们解决了传统定量和进化比较遗传学技术的固有局限性。然而,序列模型的准确性和泛化性在很大程度上取决于训练数据,这凸显了验证实验的必要性。我们指出了序列模型的成功应用,特别是在蛋白质序列方面,并确定了需要进一步改进的领域,尤其是在调控序列建模方面。虽然在计算机模拟驱动的精准育种方面还不成熟,但序列模型显示出强大的潜力,有望成为育种工具箱中不可或缺的一部分。