Shah Bipesh Kumar, Bhattarai Karun
Department of Paediatrics and Adolescent Medicine, B.P. Koirala Institute of Health Sciences, Dharan, Sunsari, Nepal.
Department of Pediatrics and Adolescents Medicine, B.P. Koirala Institute of Health Sciences, Dharan, Sunsari, Nepal.
Ann Med Surg (Lond). 2025 Jul 22;87(8):5283-5287. doi: 10.1097/MS9.0000000000003554. eCollection 2025 Aug.
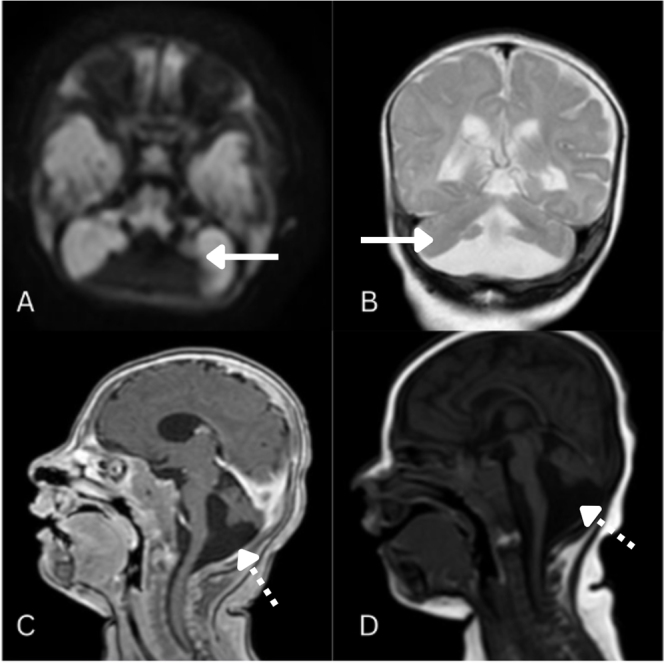
Dandy-Walker syndrome (DWS) is a rare congenital posterior fossa malformation, affecting 1 in 25 000-30 000 live births. Prenatal diagnosis is possible via ultrasound and MRI. This case highlights a rare Dandy-Walker variant with bilateral optic atrophy and status epilepticus, emphasizing the need for awareness of progressive neurological and visual impairment in Dandy-Walker spectrum disorders. The case adds to the evolving knowledge on the diverse phenotypic presentations of the Dandy-Walker variant, particularly in resource-limited settings where diagnostic and therapeutic interventions may be constrained.
A 27-month-old female with recurrent seizures since infancy presented with generalized tonic-clonic movements, ocular deviation, and frothing, lasting 2 h. Examination revealed fever, tachycardia, tachypnea, and hypertonia. MRI at 5 months confirmed Dandy-Walker spectrum disorder with cerebellar vermis hypoplasia, a posterior fossa cyst, and fourth ventricle malformation. Electroencephalogram at 14 months showed diffuse encephalopathy with multifocal seizures, and fundoscopy revealed bilateral optic atrophy. TORCH (toxoplasmosis, rubella cytomegalovirus, herpes simplex, and HIV) screening was unremarkable. Despite antiseizure therapy with levetiracetam and phenytoin, persistent seizures required pediatric intensive care unit admission. Valproate (15 mg/kg/day, titrated to 20 mg/kg/day) achieved seizure control.
This case underscores the complexity of Dandy-Walker spectrum disorder with status epilepticus in a 27-month-old female. No infectious source was identified, and lumbar puncture was declined. Seizure control required escalation of antiseizure therapy. Long-term neurodevelopmental, genetic, and neurometabolic evaluation is crucial for comprehensive management.
This case highlights the need for long-term neurodevelopmental, genetic, and neurometabolic evaluation for comprehensive management.
丹迪-沃克综合征(DWS)是一种罕见的先天性后颅窝畸形,在每25000至30000例活产中出现1例。产前诊断可通过超声和磁共振成像(MRI)实现。本病例突出了一种罕见的丹迪-沃克变异型,伴有双侧视神经萎缩和癫痫持续状态,强调了在丹迪-沃克谱系障碍中需认识到进行性神经和视力损害。该病例增加了对丹迪-沃克变异型多样表型表现的不断演变的认识,特别是在诊断和治疗干预可能受限的资源有限环境中。
一名自婴儿期起就反复发作癫痫的27个月大女性,出现全身性强直-阵挛运动、眼球偏斜和口吐白沫,持续2小时。检查发现发热、心动过速、呼吸急促和肌张力增高。5个月时的MRI证实为丹迪-沃克谱系障碍,伴有小脑蚓部发育不全、后颅窝囊肿和第四脑室畸形。14个月时的脑电图显示弥漫性脑病伴多灶性癫痫发作,眼底检查显示双侧视神经萎缩。TORCH(弓形虫病、风疹、巨细胞病毒、单纯疱疹病毒和人类免疫缺陷病毒)筛查无异常。尽管使用左乙拉西坦和苯妥英进行抗癫痫治疗,但持续性癫痫发作需要入住儿科重症监护病房。丙戊酸盐(15毫克/千克/天,滴定至20毫克/千克/天)实现了癫痫控制。
本病例强调了一名27个月大女性丹迪-沃克谱系障碍合并癫痫持续状态的复杂性。未发现感染源,且拒绝进行腰椎穿刺。癫痫控制需要加强抗癫痫治疗。长期的神经发育、遗传和神经代谢评估对于综合管理至关重要。
本病例突出了进行长期神经发育、遗传和神经代谢评估以进行综合管理的必要性。