Rodriguez-Pazmiño Angel Sebastian, Carvajal Elsy, Paredes-Núñez Darwin, Echeverría Jose, Calderon Joselyn, Orlando Solon Alberto, Parra Vera Henry, Garcia-Bereguiain Miguel Angel
One Health Research Group, Universidad de Las Américas, Quito, Ecuador.
Instituto Nacional de Salud Pública e Investigación, Guayaquil, Ecuador.
Front Cell Infect Microbiol. 2025 Jul 28;15:1612459. doi: 10.3389/fcimb.2025.1612459. eCollection 2025.
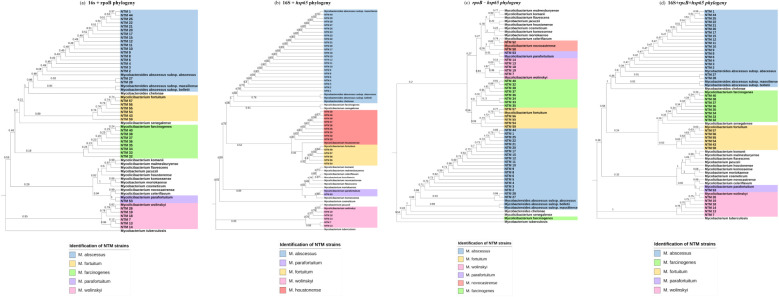
Non tuberculous mycobacteria (NTM) infections are increasing globally, underscoring the critical importance of accurate species-level identification for effective clinical management. This study aimed to evaluate the use of three conserved markers in the mycobacterial family (, , and ) for NTM identification through Sanger sequencing, comparing the results to those obtained using MALDI-ToF MS. A total of 59 clinical NTM isolates from plastic surgery patients, previously characterized by MALDI-ToF MS, were analyzed. These isolates underwent DNA extraction, PCR amplification, and Sanger sequencing. Species identification was performed through phylogenetic analyses of each marker individually and concatenated as a multi locus sequencing approach. Concordance between MALDI-ToF MS and Sanger sequencing was assessed using Cohen's Kappa statistical analysis. Cohen's Kappa values indicated moderate concordance of 0.46 for , 0.51 for , and 0.69 for . Concatenated phylogenetic analysis yielded improved concordance values of 0.71 for ( + , 0.76 for ( + , 0.69 for ( + , and 0.72 for (16S + + ). Our results show that NTM identification is more accurate when employing a multi locus sequencing approach. Notably, the combination of outperformed the three-marker concatenation, offering the highest concordance for species-level identification. NTM identification is challenging, and concatenated phylogenetic analysis of two or more gene fragments should be used when MALDI-ToF MS or whole genome sequencing is not available.
非结核分枝杆菌(NTM)感染在全球范围内呈上升趋势,这凸显了准确进行菌种水平鉴定对于有效临床管理的至关重要性。本研究旨在评估通过桑格测序法利用分枝杆菌家族中的三个保守标记(、和)进行NTM鉴定的情况,并将结果与使用基质辅助激光解吸电离飞行时间质谱(MALDI-ToF MS)获得的结果进行比较。对59株来自整形外科患者的临床NTM分离株进行了分析,这些分离株先前已通过MALDI-ToF MS进行了鉴定。对这些分离株进行了DNA提取、PCR扩增和桑格测序。通过对每个标记单独进行系统发育分析并将其串联作为多位点测序方法来进行菌种鉴定。使用科恩kappa统计分析评估MALDI-ToF MS和桑格测序之间的一致性。科恩kappa值表明,对于,一致性为0.46;对于,一致性为0.51;对于,一致性为0.69。串联系统发育分析产生了更高的一致性值,对于( + )为0.71,对于( + )为0.76,对于( + )为0.69,对于(16S + + )为0.72。我们的结果表明,采用多位点测序方法时,NTM鉴定更为准确。值得注意的是,的组合优于三个标记的串联,在菌种水平鉴定方面提供了最高的一致性。NTM鉴定具有挑战性,当无法使用MALDI-ToF MS或全基因组测序时,应使用两个或更多基因片段的串联系统发育分析。