Cerkauskaite-Kerpauskiene Agne, Navickaite Milda, Savige Judy, Mazur Gabija, Brazdziunaite Deimante, Azukaitis Karolis, Slazaite Gerda, Laurinavicius Arvydas, Miglinas Marius, Vainutiene Vija, Strupaite-Sileikiene Rasa, Misevice Ausrine, Mickeviciene Vaiva, Cerkauskiene Rimante
Vilnius University, Faculty of Medicine, Institute of Clinical Medicine, LT-03101 Vilnius, Lithuania.
Vilnius University, Faculty of Medicine, LT-03101 Vilnius, Lithuania.
Int J Mol Sci. 2025 Aug 7;26(15):7639. doi: 10.3390/ijms26157639.
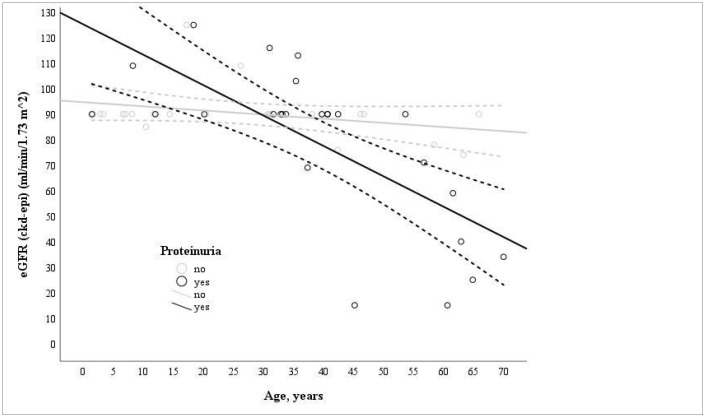
Variants in and cause autosomal dominant and recessive Alport syndrome, yet data on their distribution and clinical expression in different populations remain limited. This study investigated genotype-phenotype correlations and the distribution of / variants in a Lithuanian Alport syndrome cohort. A total of 221 individuals from Lithuania were analyzed for and variants using either next-generation sequencing or Sanger sequencing in order to assess variant distribution and associated clinical features. Only individuals with pathogenic, likely pathogenic, or uncertain significance variants were included. Fifty-two individuals (38 index cases) with pathogenic, likely pathogenic, or variants of uncertain significance were identified, as follows: forty-eight were heterozygous, four had autosomal recessive, and four had digenic Alport syndrome. variants were found in 9.5% (21/221) and in 17.6% (39/221). Among the 28 identified variants, 18 were novel. Glycine substitutions (n = 8) were the most frequent and associated with worse kidney outcomes and increased hearing abnormalities. Hematuria was diagnosed significantly earlier than proteinuria ( = 0.05). Most individuals with autosomal dominant Alport syndrome had normal kidney function (eGFR > 90 mL/min/1.73 m), while those with autosomal recessive Alport syndrome had more severe disease. Kidney failure occurred in 2/4 (50%) autosomal recessive Alport syndrome and 2/48 (4%) autosomal dominant Alport syndrome cases. A significant inverse correlation was found between eGFR and age in proteinuric individuals (r = -0.737; = 0.013). This study expands knowledge of Alport syndrome in the Lithuanian population and contributes novel variant data to the global Alport syndrome genetic database.
COL4A3和COL4A4基因的变异会导致常染色体显性和隐性阿尔波特综合征,但关于它们在不同人群中的分布和临床表型的数据仍然有限。本研究调查了立陶宛阿尔波特综合征队列中基因型与表型的相关性以及COL4A3/COL4A4基因变异的分布情况。使用二代测序或桑格测序法,对来自立陶宛的221名个体的COL4A3和COL4A4基因变异进行了分析,以评估变异分布及相关临床特征。仅纳入具有致病性、可能致病性或意义未明变异的个体。共鉴定出52名(38例索引病例)具有致病性、可能致病性或意义未明变异的个体,具体情况如下:48例为杂合子,4例为常染色体隐性遗传,4例为双基因阿尔波特综合征。在9.5%(21/221)的个体中发现了COL4A3基因变异,在17.6%(39/221)的个体中发现了COL4A4基因变异。在鉴定出的28个变异中,18个是新发现的。甘氨酸替代(n = 8)最为常见,且与更差的肾脏预后和听力异常增加相关。血尿的诊断明显早于蛋白尿(P = 0.05)。大多数常染色体显性阿尔波特综合征个体的肾功能正常(估算肾小球滤过率> 90 mL/min/1.73 m²),而常染色体隐性阿尔波特综合征个体的病情则更为严重。2/4(50%)的常染色体隐性阿尔波特综合征病例和2/48(4%)的常染色体显性阿尔波特综合征病例出现了肾衰竭。在有蛋白尿的个体中,估算肾小球滤过率与年龄之间存在显著的负相关(r = -0.737;P = 0.013)。本研究扩展了立陶宛人群中阿尔波特综合征的相关知识,并为全球阿尔波特综合征基因数据库提供了新的变异数据。