Meindl A, Hosenfeld D, Brückl W, Schuffenhauer S, Jenderny J, Bacskulin A, Oppermann H C, Swensson O, Bouloux P, Meitinger T
Abteilung für pädiatrische Genetik, Ludwig-Maximilians-Universität München, Germany.
J Med Genet. 1993 Oct;30(10):838-42. doi: 10.1136/jmg.30.10.838.
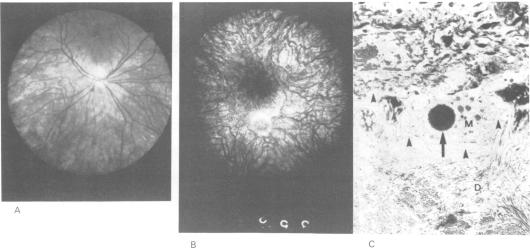
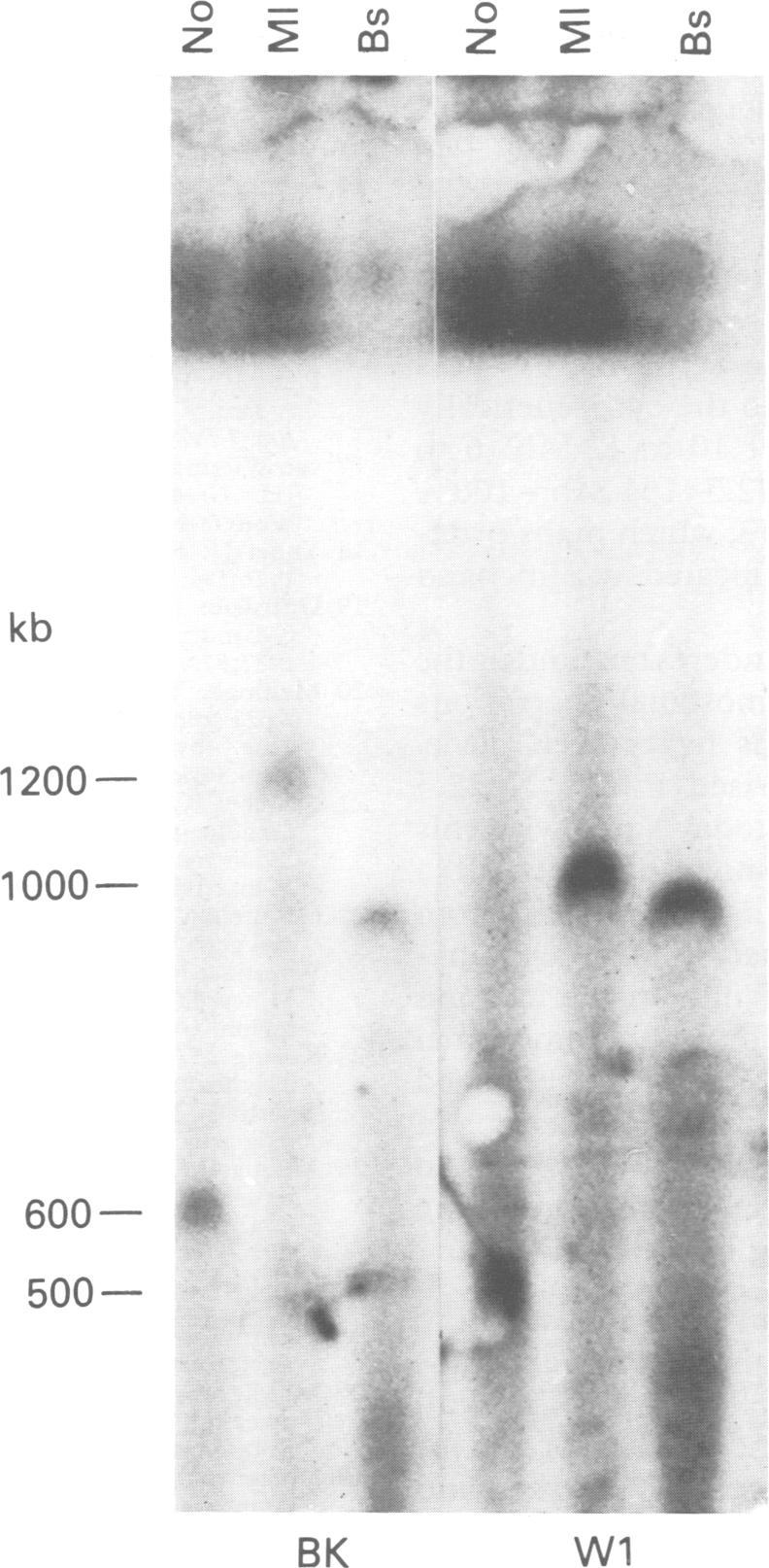
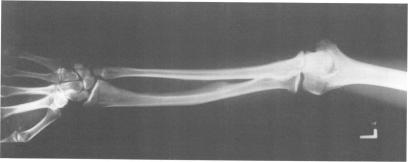
The molecular characterisation of chromosomal aberrations in Xp22.3 has established the map position of several genes with mutations resulting in diverse phenotypes such as short stature (SS), chondrodysplasia punctata (CDPX), mental retardation (MRX), ichthyosis (XLI), and Kallmann syndrome (KAL). We describe the clinical symptoms of a patient with a complex syndrome compatible with all these conditions plus ocular albinism (OA1). He has a terminal Xp deletion of at least 10 Mb of DNA. Both the mother and sister of the patient are carriers of the deletion and show a number of traits seen in Turner's syndrome. The diagnosis of ocular albinism was confirmed in the patient and his mother, who shows iris translucency, patches and streaks of hypopigmentation in the fundus, and macromelanosomes in epidermal melanocytes. By comparative deletion mapping we can define a deletion interval, which locates the OA1 gene proximal to DXS143 and distal to DXS85, with the breakpoints providing valuable starting points for cloning strategies.
对Xp22.3染色体畸变的分子特征分析已确定了几个基因的图谱位置,这些基因发生突变会导致多种不同的表型,如身材矮小(SS)、点状软骨发育不良(CDPX)、智力迟钝(MRX)、鱼鳞病(XLI)和卡尔曼综合征(KAL)。我们描述了一名患有复杂综合征患者的临床症状,该综合征与所有这些病症相符,还伴有眼白化病(OA1)。他的Xp末端缺失了至少10 Mb的DNA。患者的母亲和姐姐都是该缺失的携带者,并表现出一些特纳综合征的特征。患者及其母亲均被确诊患有眼白化病,其母亲表现为虹膜半透明、眼底有色素减退斑和条纹,以及表皮黑素细胞中有巨大黑素小体。通过比较缺失图谱分析,我们可以确定一个缺失区间,该区间将OA1基因定位在DXS143近端和DXS85远端,断点为克隆策略提供了有价值的起始点。