Lim W A, Hodel A, Sauer R T, Richards F M
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511.
Proc Natl Acad Sci U S A. 1994 Jan 4;91(1):423-7. doi: 10.1073/pnas.91.1.423.
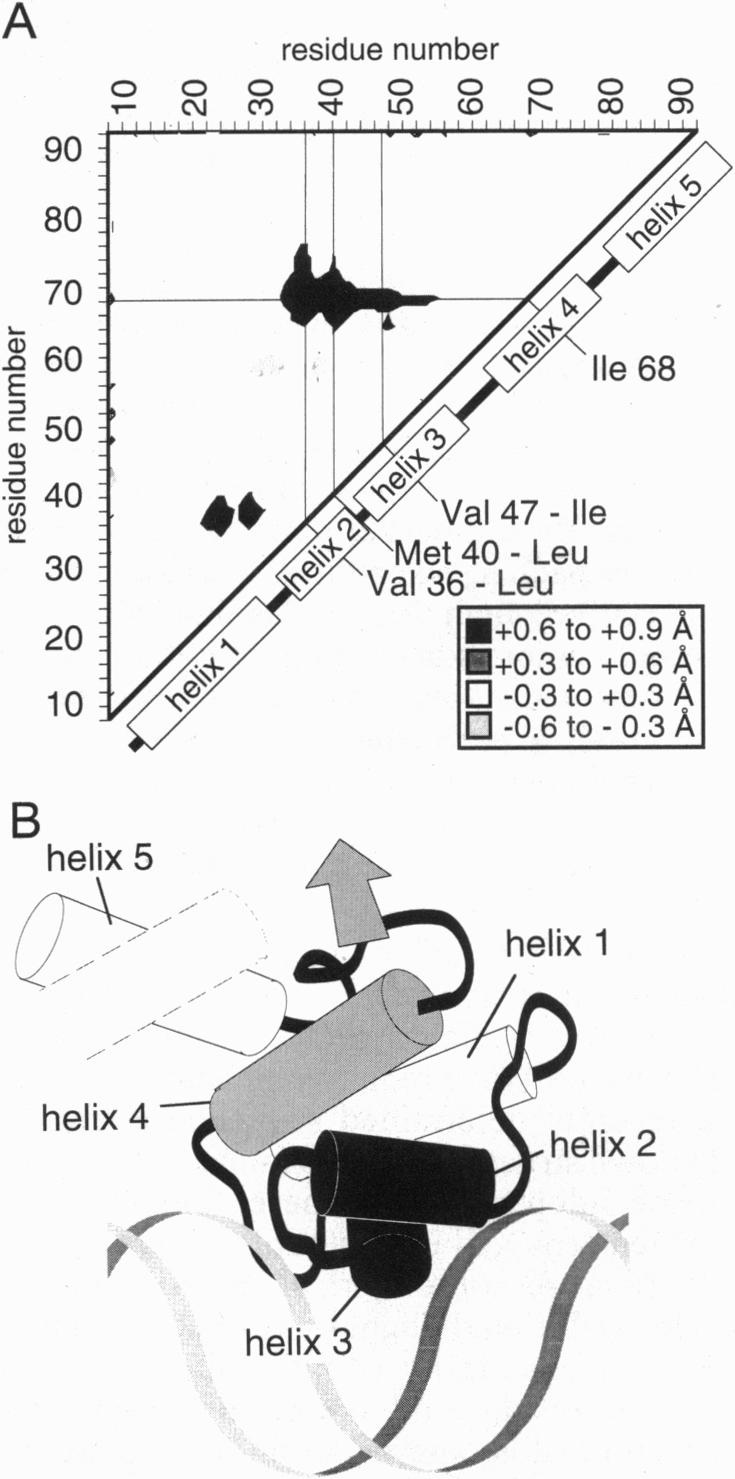
The dense packing observed in protein interiors appears to be crucial for stabilizing the native structure--even subtle internal substitutions are usually destabilizing. Thus, steric complementarity of core residues is thought to be an important criterion for "inverse folding" predictive methods, which judge whether a newly determined sequence is consistent with any known folds. A major problem in the development of useful core packing evaluation algorithms, however, is that there are occasional mutations that are predicted to disrupt native packing but that yield an equally or more stable protein. We have solved the crystal structure of such a variant of lambda repressor, which, despite having three larger core substitutions, is more stable than the wild type. The structure reveals that the protein accommodates the potentially disruptive residues with shifts in its alpha-helical arrangement. The variant is apparently more stable because its packing is improved--the core has a higher packing density and little geometric strain. These rearrangements, however, cause repositioning of functional residues, which result in reduced DNA binding activity. By comparing these results with the predictions of two core packing algorithms, it is clear that the protein possesses a relatively high degree of main-chain flexibility that must be accounted for in order to predict the full spectrum of compatible core sequences. This study also shows how, in protein evolution, a particular set of core residue identities might be selected not because they provide optimal stability but because they provide sufficient stability in addition to the precise structure required for optimal activity.
在蛋白质内部观察到的紧密堆积对于稳定天然结构似乎至关重要——即使是微小的内部替换通常也会导致结构不稳定。因此,核心残基的空间互补性被认为是“反向折叠”预测方法的一个重要标准,该方法用于判断新确定的序列是否与任何已知折叠结构一致。然而,开发有用的核心堆积评估算法的一个主要问题是,偶尔会出现一些突变,这些突变预计会破坏天然堆积,但却能产生同样稳定或更稳定的蛋白质。我们解析了λ阻遏物这种变体的晶体结构,尽管它有三个较大的核心替换,但比野生型更稳定。该结构表明,蛋白质通过其α螺旋排列的改变来容纳潜在的破坏性残基。该变体显然更稳定,因为其堆积得到了改善——核心具有更高的堆积密度且几乎没有几何应变。然而,这些重排导致了功能残基的重新定位,从而导致DNA结合活性降低。通过将这些结果与两种核心堆积算法的预测结果进行比较,很明显该蛋白质具有相对较高程度的主链灵活性,为了预测兼容核心序列的完整范围,必须考虑这一点。这项研究还表明,在蛋白质进化过程中,选择特定的一组核心残基身份可能不是因为它们提供了最佳稳定性,而是因为它们除了提供最佳活性所需的精确结构外,还提供了足够的稳定性。