Feussner G
Department of Internal Medicine I, Endocrinology and Metabolism, University of Heidelberg, Germany.
J Clin Pathol. 1996 Dec;49(12):985-9. doi: 10.1136/jcp.49.12.985.
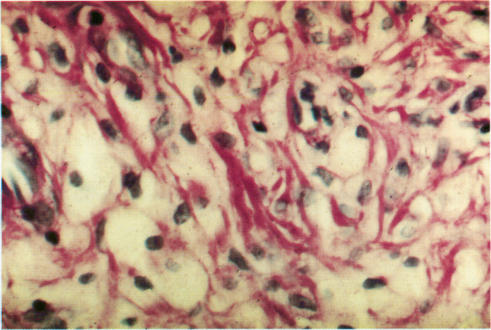
To present the clinical, dermatological, and histological features of a patient with generalised xanthomatosis, familial apolipoprotein (apo) E deficiency, and unusual type III hyperlipoproteinaemia (HLP).
The underlying molecular defect was disclosed using molecular biological techniques. The unusual xanthomas were histologically analysed and the morphology of the abnormal lipoprotein particles examined using electron microscopy.
A 10 base pair deletion in exon 4 of the proband's apo epsilon gene (base pairs 4037-4046 coding for amino acids 209-212 of the mature protein) was identified. This is predictive for a reading frameshift encoding a premature stop (TGA) in codon 229. The mutation is responsible for delayed catabolism of atherogenic lipoprotein remnants, lipid storage in monocyte/macrophages, and phenotypic expression of xanthomatosis early in life.
Familial apo E deficiency is a rare genetic disease which offers the unique opportunity to study the impact of apo E on lipoprotein metabolism and development of atherosclerosis in humans.
介绍一名患有全身性黄瘤病、家族性载脂蛋白(apo)E缺乏症和不寻常的III型高脂蛋白血症(HLP)患者的临床、皮肤和组织学特征。
使用分子生物学技术揭示潜在的分子缺陷。对不寻常的黄瘤进行组织学分析,并使用电子显微镜检查异常脂蛋白颗粒的形态。
在先证者的载脂蛋白ε基因第4外显子中发现了一个10个碱基对的缺失(碱基对4037 - 4046编码成熟蛋白的氨基酸209 - 212)。这预示着编码第229密码子中过早终止(TGA)的读框移位。该突变导致致动脉粥样硬化脂蛋白残粒的分解代谢延迟、单核细胞/巨噬细胞中的脂质储存以及早年黄瘤病的表型表达。
家族性载脂蛋白E缺乏症是一种罕见的遗传疾病,为研究载脂蛋白E对人类脂蛋白代谢和动脉粥样硬化发展的影响提供了独特的机会。