Lühder F, Katz J, Benoist C, Mathis D
Institut de Génétique et de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique/Institut National de la Santé et de la Recherche Médicale/Université Louis Pasteur, 67404 Illkirch, Strasbourg, France.
J Exp Med. 1998 Feb 2;187(3):379-87. doi: 10.1084/jem.187.3.379.
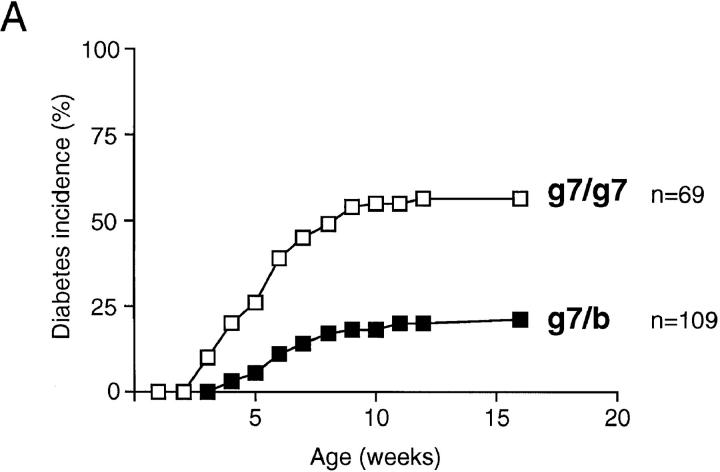
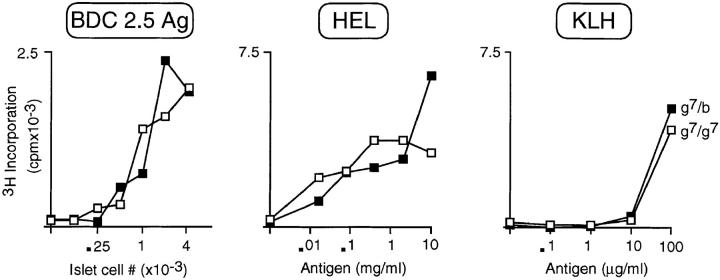
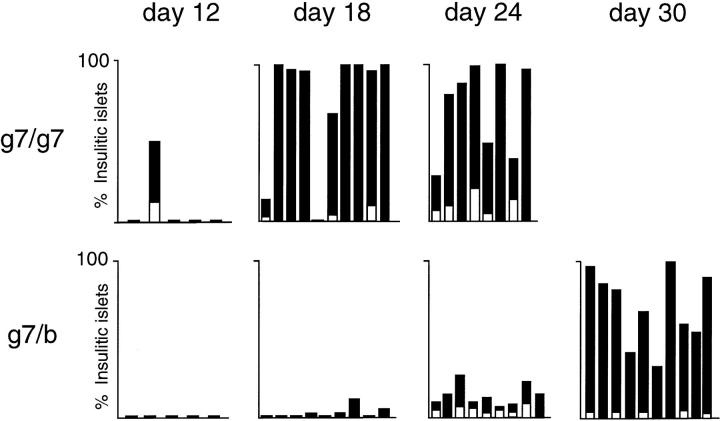
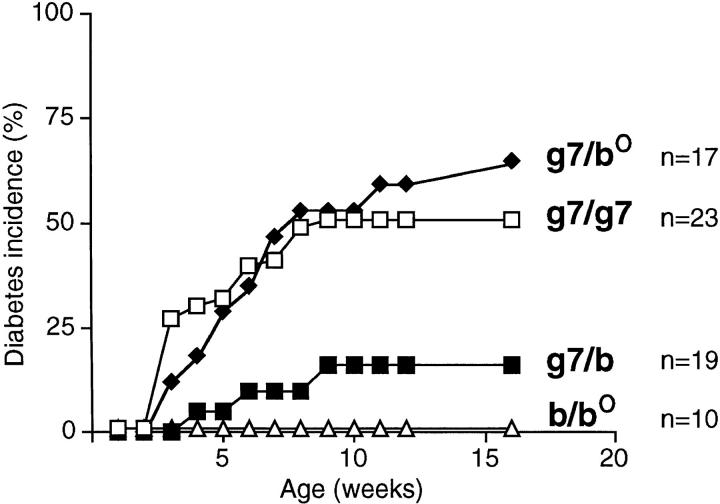
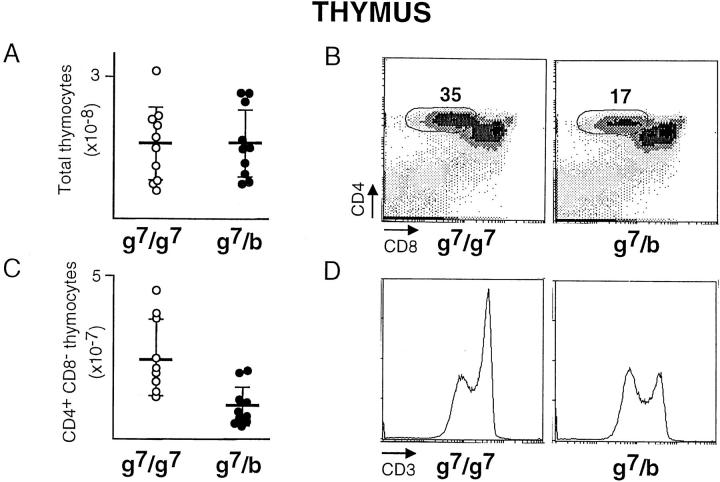
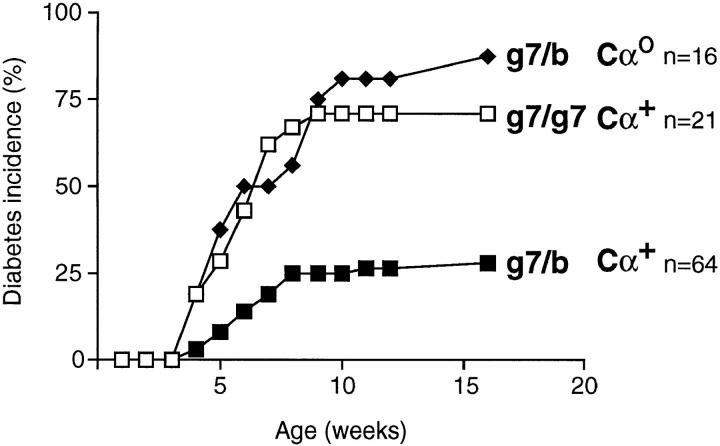
Insulin-dependent diabetes is heavily influenced by genes encoded within the major histocompatibility complex (MHC), positively by some class II alleles and negatively by others. We have explored the mechanism of MHC class II-mediated protection from diabetes using a mouse model carrying the rearranged T cell receptor (TCR) transgenes from a diabetogenic T cell clone derived from a nonobese diabetic mouse. BDC2.5 TCR transgenics with C57Bl/6 background genes and two doses of the H-2(g7) allele exhibited strong insulitis at approximately 3 wk of age and most developed diabetes a few weeks later. When one of the H-2(g7) alleles was replaced by H-2(b), insulitis was still severe and only slightly delayed, but diabetes was markedly inhibited in both its penetrance and time of onset. The protective effect was mediated by the Abetab gene, and did not merely reflect haplozygosity of the Abetag7 gene. The only differences we observed in the T cell compartments of g7/g7 and g7/b mice were a decrease in CD4(+) cells displaying the transgene-encoded TCR and an increase in cells expressing endogenously encoded TCR alpha-chains. When the synthesis of endogenously encoded alpha-chains was prevented, the g7/b animals were no longer protected from diabetes. g7/b mice did not have a general defect in the production of Ag7-restricted T cells, and antigen-presenting cells from g7/b animals were as effective as those from g7/g7 mice in stimulating Ag7-restricted T cell hybridomas. These results argue against mechanisms of protection involving clonal deletion or anergization of diabetogenic T cells, or one depending on capture of potentially pathogenic Ag7-restricted epitopes by Ab molecules. Rather, they support a mechanism based on MHC class II-mediated positive selection of T cells expressing additional specificities.
胰岛素依赖型糖尿病受到主要组织相容性复合体(MHC)内编码基因的严重影响,一些II类等位基因起正向作用,而另一些则起负向作用。我们利用一种小鼠模型探讨了MHC II类介导的预防糖尿病的机制,该小鼠模型携带来自非肥胖糖尿病小鼠的致糖尿病T细胞克隆的重排T细胞受体(TCR)转基因。具有C57Bl/6背景基因和两剂H-2(g7)等位基因的BDC2.5 TCR转基因小鼠在约3周龄时表现出强烈的胰岛炎,几周后大多数发展为糖尿病。当其中一个H-2(g7)等位基因被H-2(b)取代时,胰岛炎仍然严重且仅略有延迟,但糖尿病的发病率和发病时间均明显受到抑制。这种保护作用由Abetab基因介导,并非仅仅反映Abetag7基因的单倍体状态。我们在g7/g7和g7/b小鼠的T细胞区室中观察到的唯一差异是,显示转基因编码TCR的CD4(+)细胞减少,而表达内源性编码TCRα链的细胞增加。当内源性编码α链的合成被阻止时,g7/b动物不再受到糖尿病的保护。g7/b小鼠在产生Ag7限制性T细胞方面没有普遍缺陷,并且来自g7/b动物的抗原呈递细胞在刺激Ag7限制性T细胞杂交瘤方面与来自g7/g7小鼠的细胞一样有效。这些结果反对涉及致糖尿病T细胞的克隆缺失或失能的保护机制,也反对一种依赖于Ab分子捕获潜在致病性Ag7限制性表位的机制。相反,它们支持一种基于MHC II类介导的对表达其他特异性的T细胞进行阳性选择的机制。