Sjarif D R, Sinke R J, Duran M, Beemer F A, Kleijer W J, Ploos van Amstel J K, Poll-The B T
University Children's Hospital Het Wilhelmina Kinderziekenhuis, Utrecht, The Netherlands.
J Med Genet. 1998 Aug;35(8):650-6. doi: 10.1136/jmg.35.8.650.
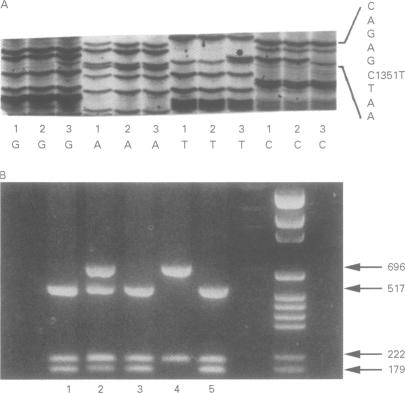
Isolated glycerol kinase deficiency (GKD) is an X linked recessive disorder. The clinical and biochemical picture may vary from a childhood metabolic crisis to asymptomatic adult "pseudohypertriglyceridaemia", the result of hyperglycerolaemia. We performed glycerol kinase (GK) gene analysis to study the molecular heterogeneity and genotype-phenotype correlation in eight males from three families with isolated GKD. All patients had hyperglycerolaemia and glyceroluria. Four patients from two families were essentially free of symptoms. Three patients had gastrointestinal symptoms with ketoacidosis or hypoglycaemia or both. One patient had recurrent convulsions as the only acute sign, without evidence that it was correlated with a catabolic state. Fasting tests in two symptomatic patients of family 1 showed hyperketotic states, together with a tendency to hypoglycaemia. The diagnosis was confirmed by a defective 14C-glycerol incorporation into trichloroacetic acid precipitable macromolecules in intact skin fibroblasts. Mutation screening of the GK gene was performed by amplification and direct sequencing of exons using PCR. Three novel mutations were identified: (1) a deletion starting downstream of exon 9, extending to the 3' end of the gene; (2) a nonsense mutation R413X caused by a C1351T transition; and (3) a missense mutation W503R caused by a T1651C transition. In addition, we found differences from the reported sequence: (1) exon 9 actually consists of two exons, which consequently will change the number of GK gene exons from 19 to 20 exons, and (2) nucleotide differences in exon 19. So far, no genotype-phenotype correlation can be established in these GKD families.
孤立性甘油激酶缺乏症(GKD)是一种X连锁隐性疾病。其临床和生化表现可能各不相同,从儿童期的代谢危机到无症状的成人“假性高甘油三酯血症”(高甘油血症的结果)。我们对来自三个患有孤立性GKD家庭的八名男性进行了甘油激酶(GK)基因分析,以研究分子异质性和基因型 - 表型相关性。所有患者均有高甘油血症和甘油尿症。来自两个家庭的四名患者基本无症状。三名患者有胃肠道症状,伴有酮症酸中毒或低血糖症或两者皆有。一名患者以反复惊厥为唯一急性症状,无证据表明其与分解代谢状态相关。对家族1的两名有症状患者进行的禁食试验显示有酮症状态,同时有低血糖倾向。通过完整皮肤成纤维细胞中14C - 甘油掺入三氯乙酸可沉淀大分子的缺陷来确诊。使用PCR对外显子进行扩增和直接测序,对GK基因进行突变筛查。鉴定出三个新突变:(1)一个从外显子9下游开始的缺失,延伸至基因的3'端;(2)由C1351T转换引起的无义突变R413X;(3)由T1651C转换引起的错义突变W503R。此外,我们发现与报道序列存在差异:(1)外显子9实际上由两个外显子组成,因此GK基因外显子数量将从19个变为20个,以及(2)外显子19中的核苷酸差异。到目前为止,在这些GKD家族中尚未建立基因型 - 表型相关性。