JCI新发现:生酮作用不仅是“燃脂”,更是阻断脂肪肝恶化的关键防线
学术资讯

在这个代谢疾病日益高发的时代,脂肪肝(MASLD,代谢功能障碍相关脂肪性肝病)正悄然成为全球健康的隐形重负。这不仅是一个关于“肥胖”的话题,更是一个复杂的细胞代谢谜题。传统观念中,我们倾向于认为脂肪肝的恶化仅仅是因为肝脏“燃烧”脂肪(氧化)的速度赶不上脂肪堆积的速度。然而,发表在权威医学期刊《临床研究杂志》(The Journal of Clinical Investigation)上的一项最新研究颠覆了这一线性认知:肝脏的生酮作用(Ketogenesis)不仅仅是能量代谢的副产物,它本身可能就是防止肝脏走向纤维化和严重损伤的关键保护机制,且其作用原理远超简单的“脂肪氧化”范畴。
为了解开从脂肪肝(MASLD)向脂肪性肝炎(MASH)恶化的代谢密码,来自明尼苏达大学医学院的研究团队并没有仅仅停留在动物模型上,而是首先深入探究了人类患者的真实代谢状态。研究人员招募了一组经组织学确诊的MASH患者,通过先进的核磁共振(NMR)光谱和稳定同位素示踪技术,对他们体内的代谢流进行了精确量化。
这就好比在繁忙的城市交通网中,不仅仅统计有多少车(脂肪)进入了城市,还通过“GPS定位”(同位素示踪)精确追踪了每一辆车是走了高速公路(三羧酸循环氧化)还是走了高架桥(生酮作用)。
如图[1]所示,研究团队构建了一个涵盖从单纯性脂肪肝到脂肪性肝炎再到肝硬化的疾病谱系模型。通过对患者进行肝活检、葡萄糖耐量测试以及详细的代谢流分析,研究人员试图寻找代谢灵活性与肝脏病理评分(NAS,非酒精性脂肪肝活动度评分)之间的联系。
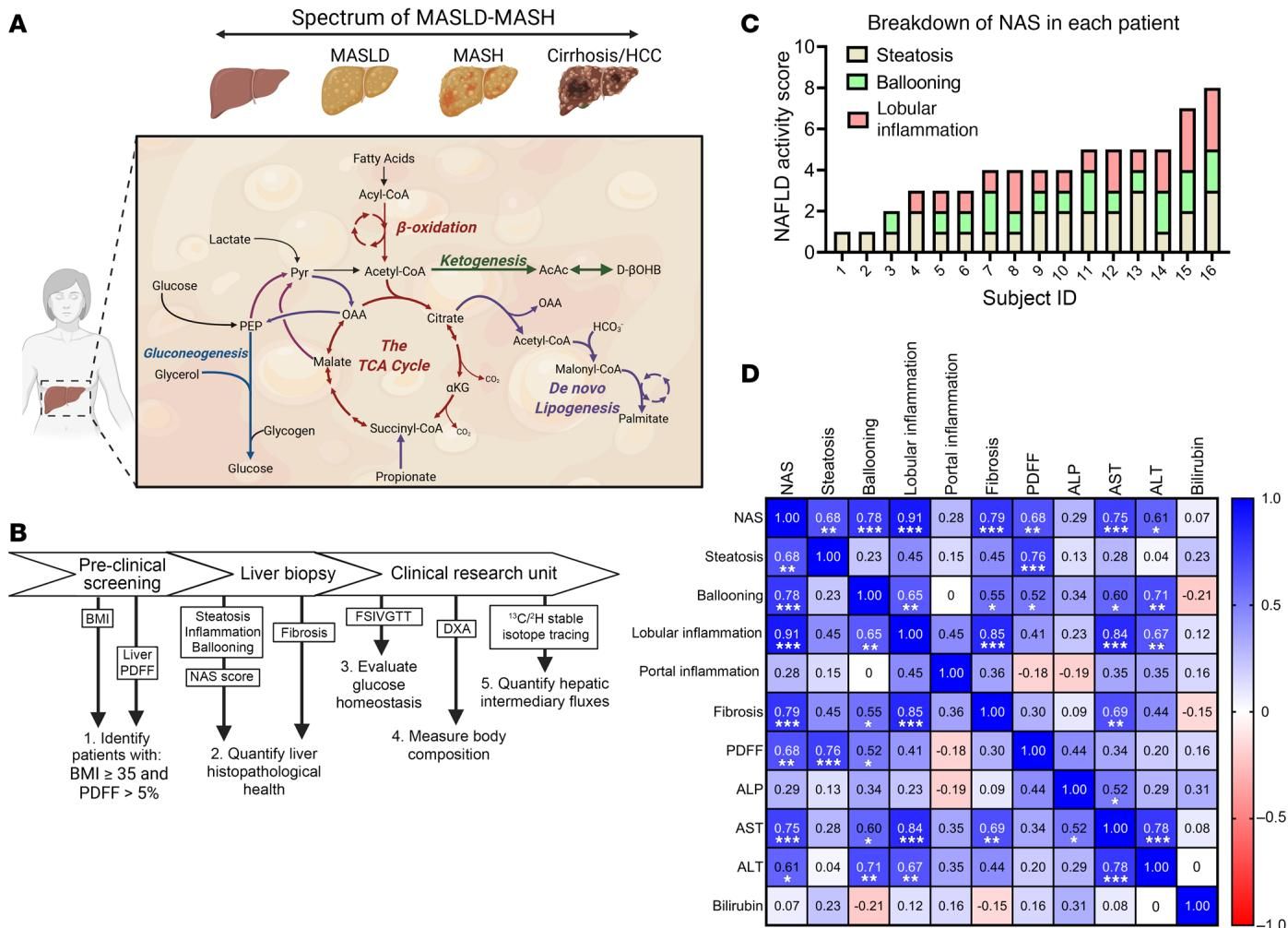
令人意外的结果出现了:肝脏损伤越严重的患者,其生酮作用反而越活跃。
传统观点可能认为,患病的肝脏代谢功能应该全面衰退。但数据显示,虽然患者的胰岛素抵抗与肝损伤评分相关,但在代谢流层面,肝脏损伤程度(NAS评分)与内源性生酮速率()呈现出显著的正相关。如图[2]所示,从相关性分析中我们可以清晰地看到,随着NAS评分(横轴)的增加,患者的生酮速率(纵轴A)和总脂肪氧化率(纵轴B)都在显著上升。然而,作为细胞能量工厂核心的三羧酸循环(TCA Cycle)周转率(纵轴F)却与肝损伤程度无关。
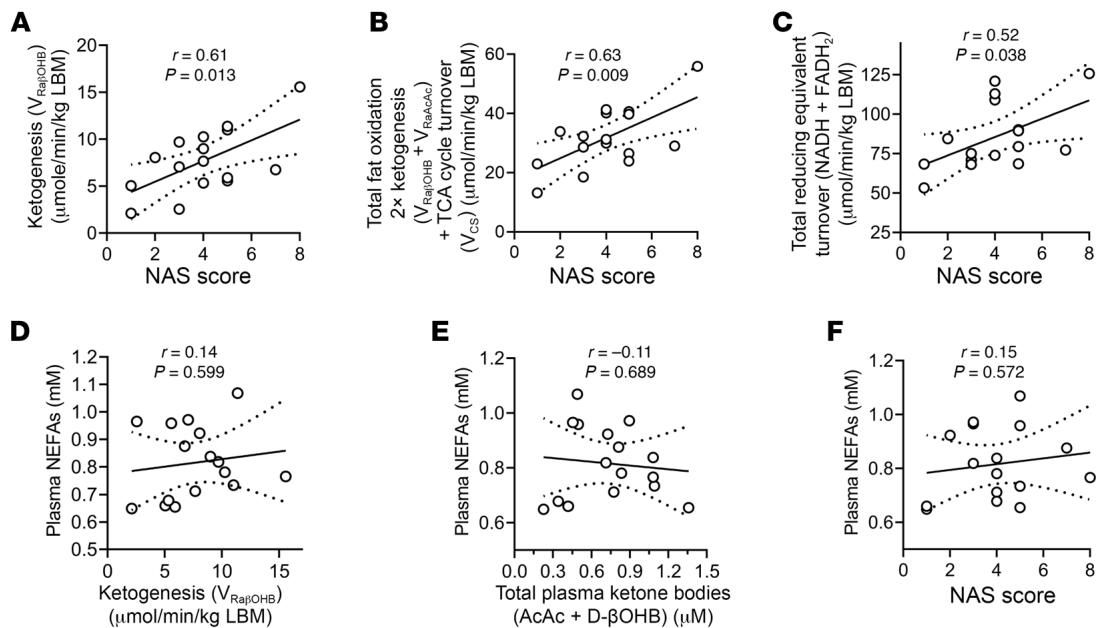
这一发现制造了一个巨大的认知冲突:如果肝脏已经在努力“燃烧”更多的脂肪并产生酮体,为什么病情还在恶化?这种生酮作用的增强,究竟是导致损伤的“凶手”,还是肝脏在绝境中启动的“代偿性自救”?
为了厘清生酮作用究竟是敌是友,研究团队将目光转向了基因编辑小鼠模型。他们构建了肝脏特异性敲除 Hmgcs2 基因的小鼠(HMGCS2-Liver-KO)。HMGCS2 是线粒体中生酮作用的限速酶,敲除它就等于人为切断了肝脏生成酮体的能力。
实验结果极具视觉冲击力。当这些无法产生酮体的小鼠被喂食高脂饮食(HFCR)一周后,它们的体重虽然下降了,但肝脏却发生了惊人的变化。如图[3]所示,与野生型(WT)小鼠相比,敲除了生酮关键酶的小鼠(HMGCS2 KO)肝脏重量显著增加(图B、C),并且出现了严重的肝肿大。更直观的是图E中的组织切片对比,HMGCS2 KO小鼠的肝脏细胞中充满了密密麻麻的脂滴,显示出极度严重的脂肪变性。
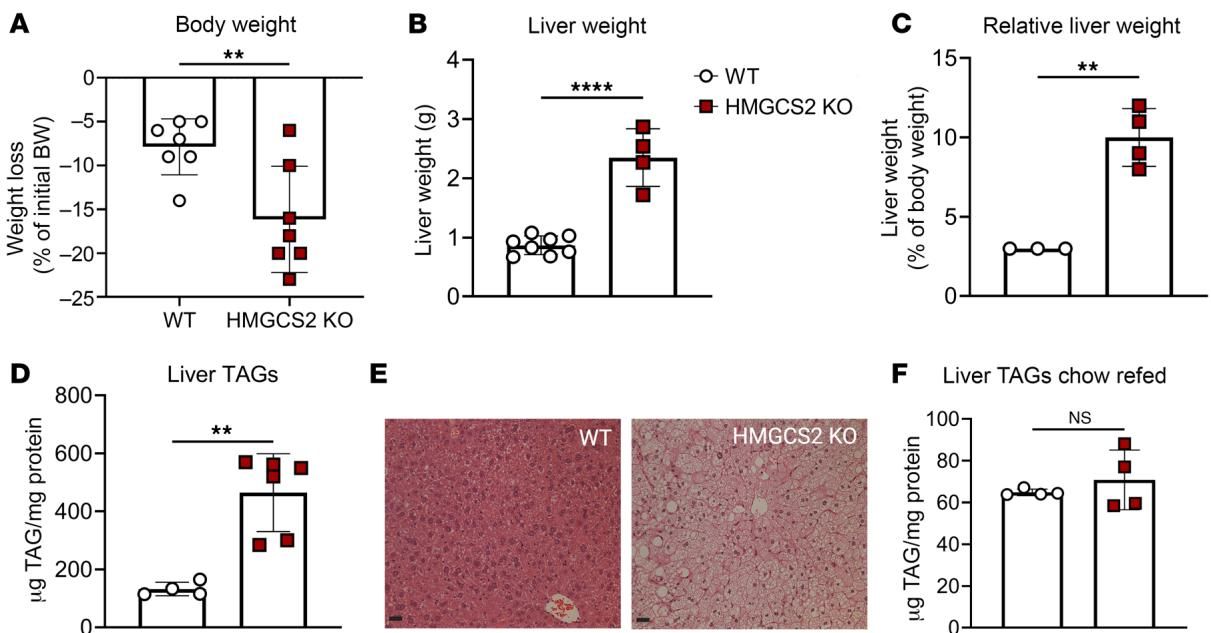
这一结果有力地证明了:生酮作用是维持肝脏脂肪稳态的“必需品”。 一旦生酮途径被阻断,即便是在同等饮食条件下,肝脏处理脂肪的能力也会迅速崩溃,导致脂质在肝细胞内疯狂堆积。
那么,这背后的微观机制是什么?是因为切断生酮导致脂肪“烧”不动了吗?
研究人员利用线粒体诊断平台(Mitochondrial diagnostics)对这一过程进行了剖析。如图[4]所示,通过提取小鼠肝脏线粒体并进行代谢流分析,研究发现,当使用HMGCS抑制剂(HG)处理线粒体模拟生酮缺陷时,线粒体的呼吸功能受到显著抑制。具体表现为,在能量需求增加时,呼吸速率的上升斜率变缓(图D),这意味着线粒体“喘不上气”了。同时,酰基辅酶A(Acyl-CoA)衍生物大量堆积(图E),而游离辅酶A(Free CoA)却被耗尽(图F)。
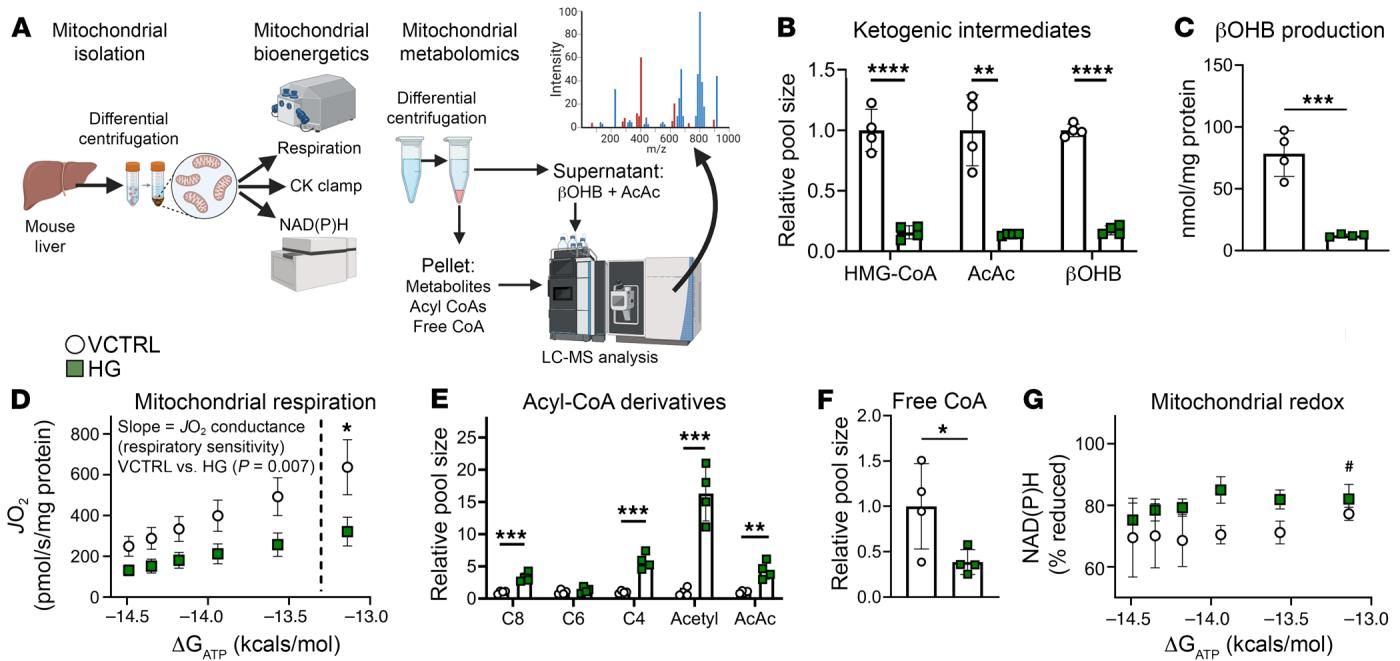
这揭示了一个核心机制:生酮作用不仅仅是产生酮体,它更像是一个“解压阀”,通过释放游离辅酶A,防止代谢中间产物堵塞线粒体,从而保证脂肪氧化(-oxidation)的顺畅进行。失去了这个解压阀,脂肪氧化的齿轮就被卡住了。
如果故事到这里结束,结论似乎很明确:生酮作用通过维持脂肪氧化来保护肝脏。然而,科学探索的魅力往往在于那些意料之外的“反转”。研究团队为了验证这一假设,构建了另一个小鼠模型——肝脏特异性敲除 Bdh1 基因的小鼠(BDH1-Liver-KO)。
BDH1(-羟基丁酸脱氢酶)负责生酮通路的最后一步,将乙酰乙酸(AcAc)转化为我们熟知的-羟基丁酸(OHB),同时这一过程也是线粒体再生NAD+的重要途径。理论上,敲除BDH1也会像敲除HMGCS2一样,破坏生酮通路的完整性,进而影响脂肪氧化。
代谢流分析证实了这一点。如图[5]所示,BDH1敲除小鼠(蓝色柱)的肝脏确实出现了代谢异常。与野生型相比,它们的三羧酸循环周转率(图D中的)和总脂肪氧化率(图E)均显著下降。这意味着,从“燃烧脂肪”的角度看,BDH1敲除小鼠和HMGCS2敲除小鼠一样,都经历了脂肪氧化能力的“减速”。
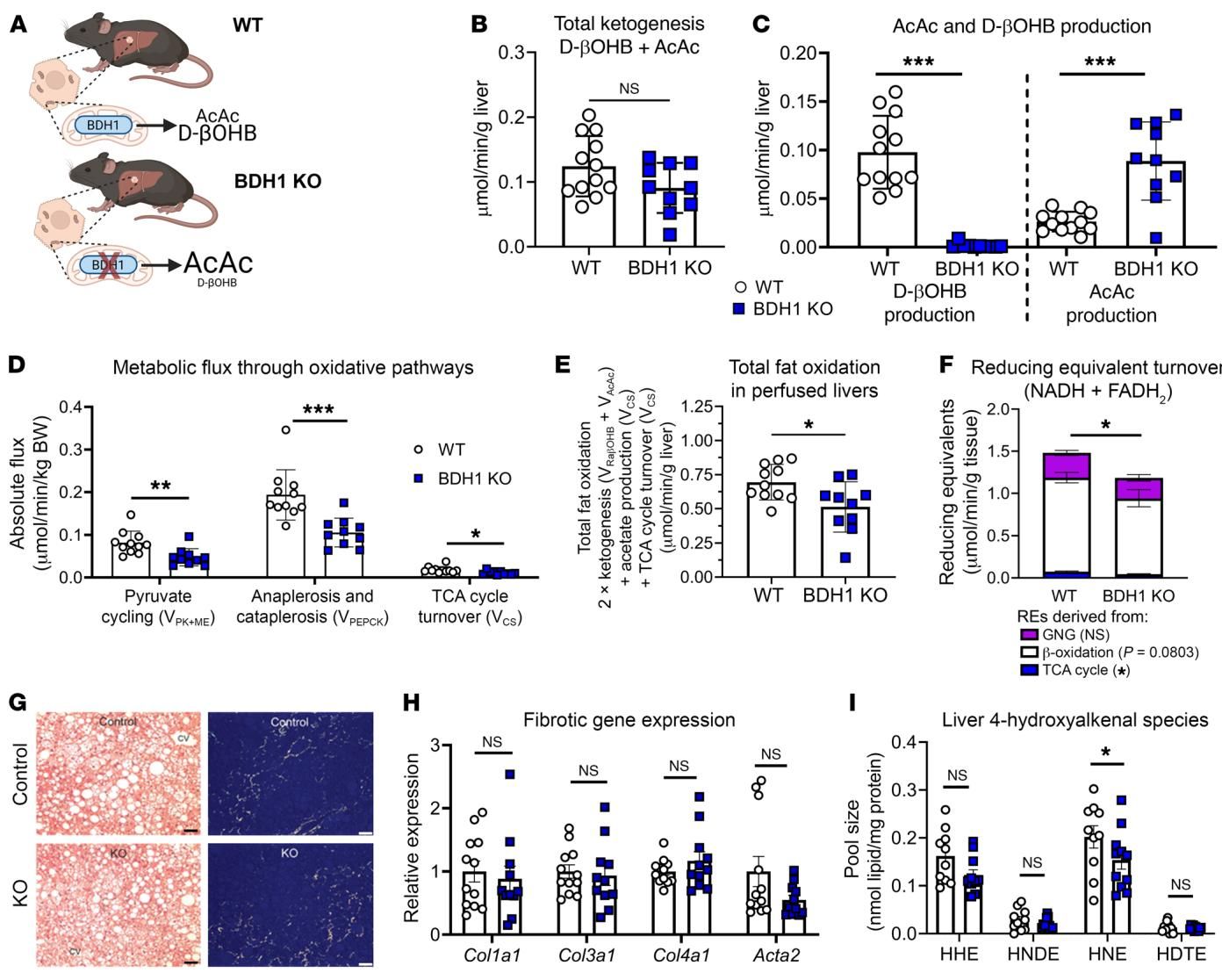
按照之前的逻辑,我们理应看到BDH1敲除小鼠的肝脏也像HMGCS2敲除小鼠那样,充斥着脂肪甚至出现纤维化。但令人大跌眼镜的是,这种情况并没有发生。
如图[5](G-I)所示,在长期喂食高脂饮食的诱导下,BDH1敲除小鼠的肝脏并没有表现出更严重的纤维化(Picrosirius red染色未见明显差异),纤维化相关的基因表达(如 Col1a1, Acta2)也没有显著升高,甚至脂质过氧化产物(HNE)的水平也相对稳定。换句话说,尽管脂肪氧化受损了,但肝脏似乎“挺住了”,并没有滑向严重的病理深渊。
这两个小鼠模型的鲜明对比,为我们揭示了一个深层次的科学事实:脂肪氧化率的降低(Fat Oxidation Rate),并不是导致脂肪肝恶化为脂肪性肝炎(MASH)的唯一或决定性因素。
这暗示了生酮作用的保护机制远比简单的“能量燃烧”更为复杂。一种可能的解释是,生酮通路中的特定中间产物(如乙酰乙酸)或其对细胞内环境(如氧化还原状态、辅酶A池的循环)的调节,发挥了独立于“燃烧卡路里”之外的保护作用。例如,研究团队在讨论中提到,生酮作用可能与肝脏中多不饱和脂肪酸(PUFA)的合成密切相关,而PUFA具有抗炎和抑制脂肪生成的特性;此外,乙酰乙酸本身也被发现具有抗纤维化的信号功能。
回到人类患者身上,我们在开篇提到的“肝损伤越重,生酮越强”的现象,现在有了更合理的解释:这很可能是肝脏在面临脂质过载和炎症压力时,启动的一种代偿性保护机制。肝脏试图通过加速生酮来释放被囚禁的辅酶A,维持线粒体功能,并产生具有细胞保护作用的酮体分子。这种升高不是致病的原因,而是身体顽强抵抗疾病留下的痕迹。
这项发表于 JCI 的研究重塑了我们对脂肪肝代谢机制的理解。它提醒我们,在开发针对MASH的药物时,简单粗暴地追求“提高脂肪氧化率”可能并不足以解决问题,甚至可能南辕北辙。如果一种药物仅仅促进了脂肪进入线粒体氧化,却忽略了生酮通路的顺畅(如辅酶A的解离和循环),可能会导致代谢中间产物的堆积,反而加重线粒体压力。
未来的治疗策略或许应该更精细地关注如何维护生酮通路的完整性,或者探索酮体分子本身作为“代谢信号”在抗纤维化中的治疗潜力。在这个微观的代谢战场上,生酮作用不再仅仅是饥饿时的备用发电机,它是肝脏抵御脂毒性风暴的一道关键防线,值得我们给予更多的关注与敬畏。
本文由超能文献“资讯AI智能体”基于4000万篇Pubmed文献自主选题与撰写,并经AI核查及编辑团队二次人工审校。内容仅供学术交流参考,不代表任何医学建议。
分享

STTT顶刊揭示:陆军军医大学吴玉章教授团队研究发现,脂肪细胞并非炎症受害者,而是幕后黑手。肥胖时,脂肪细胞过量表达B2M蛋白,既激活CD8+ T细胞,又引发自身“铁死亡”,双重机制引爆全身慢性炎症。靶向B2M有望逆转肥胖及代谢并发症。

在Zotero中直接翻译PDF文献,提供准确的学术翻译,助力您快速理解外文资料,提升文献处理效率。无缝集成,一键操作,是研究者的得力助手


荷兰瓦赫宁根大学JAMA Open研究追踪1751名结直肠癌患者5年,发现膳食纤维显著降低术后腹泻风险56%,尤其在化疗和放疗幸存者中效果显著,同时改善生活质量。