Terwilliger T C
Bioscience Division, Mail Stop M888, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
Acta Crystallogr D Biol Crystallogr. 2001 Dec;57(Pt 12):1763-75. doi: 10.1107/s0907444901013749. Epub 2001 Nov 21.
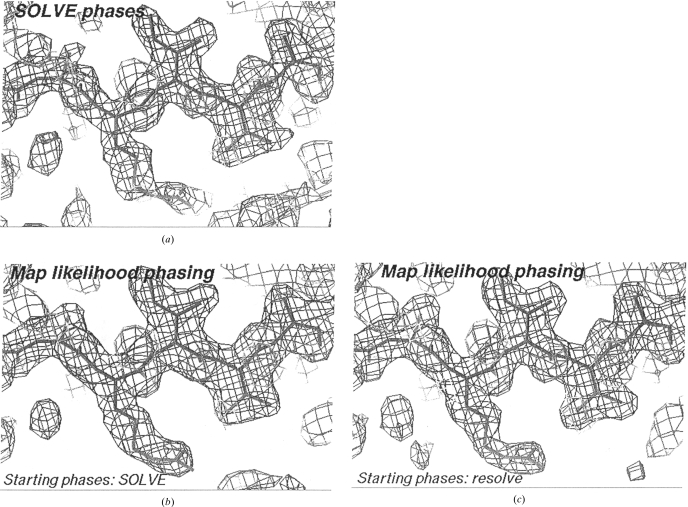
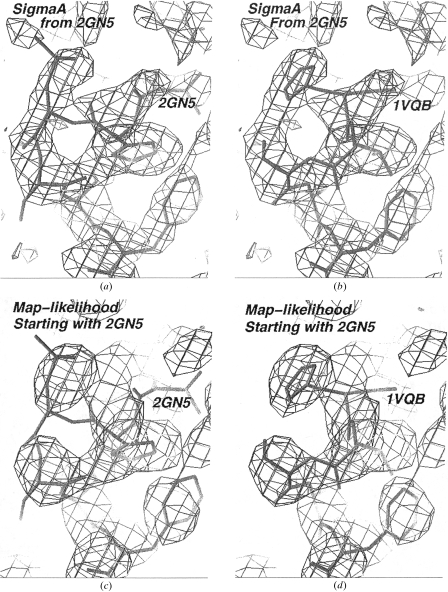
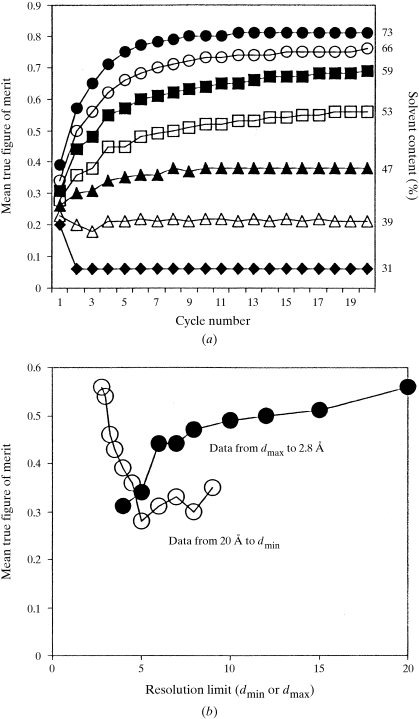
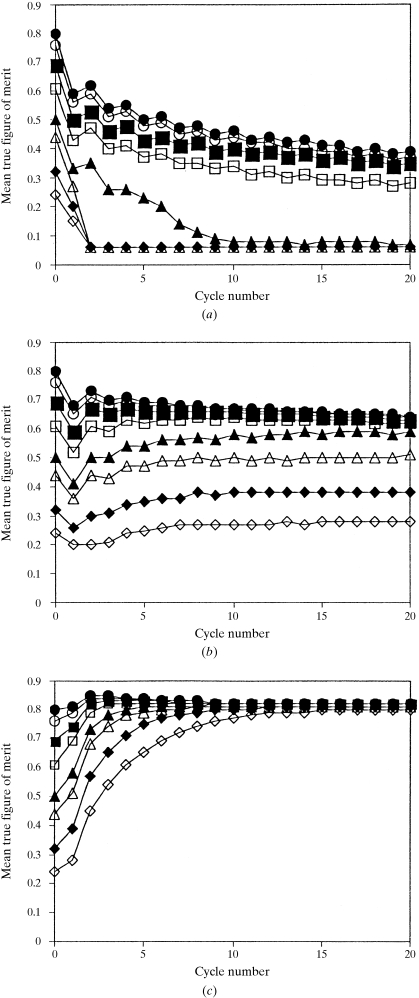
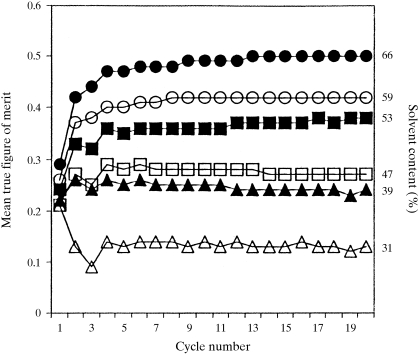
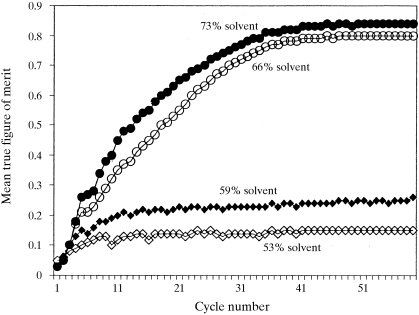
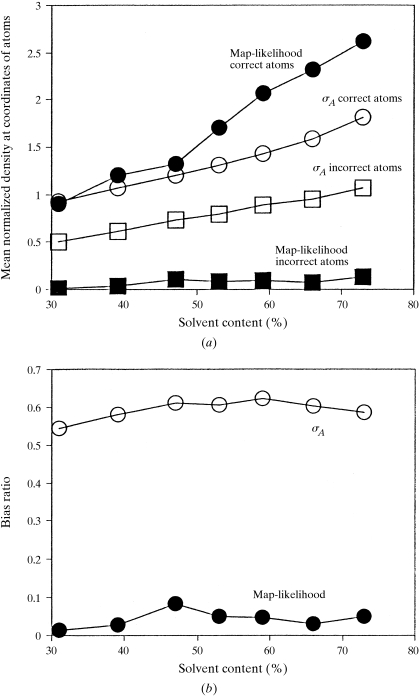
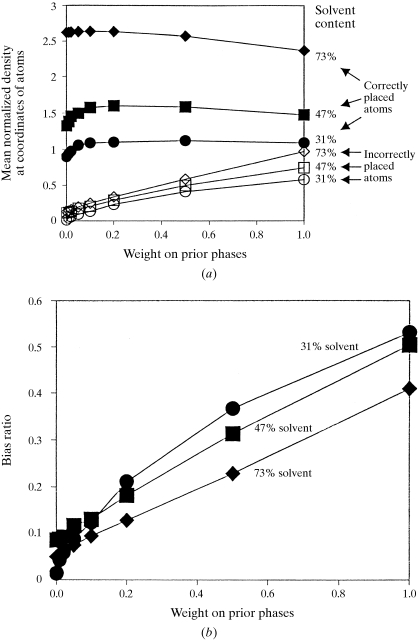
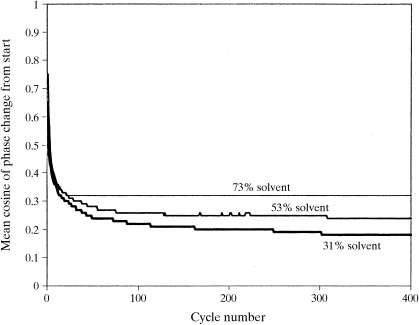
The recently developed technique of maximum-likelihood density modification [Terwilliger (2000), Acta Cryst. D56, 965-972] allows a calculation of phase probabilities based on the likelihood of the electron-density map to be carried out separately from the calculation of any prior phase probabilities. Here, it is shown that phase-probability distributions calculated from the map-likelihood function alone can be highly accurate and that they show minimal bias towards the phases used to initiate the calculation. Map-likelihood phase probabilities depend upon expected characteristics of the electron-density map, such as a defined solvent region and expected electron-density distributions within the solvent region and the region occupied by a macromolecule. In the simplest case, map-likelihood phase-probability distributions are largely based on the flatness of the solvent region. Though map-likelihood phases can be calculated without prior phase information, they are greatly enhanced by high-quality starting phases. This leads to the technique of prime-and-switch phasing for removing model bias. In prime-and-switch phasing, biased phases such as those from a model are used to prime or initiate map-likelihood phasing, then final phases are obtained from map-likelihood phasing alone. Map-likelihood phasing can be applied in cases with solvent content as low as 30%. Potential applications of map-likelihood phasing include unbiased phase calculation from molecular-replacement models, iterative model building, unbiased electron-density maps for cases where 2F(o) - F(c) or sigma(A)-weighted maps would currently be used, structure validation and ab initio phase determination from solvent masks, non-crystallographic symmetry or other knowledge about expected electron density.
最近开发的最大似然密度修正技术[特威利格(2000年),《晶体学报》D56卷,965 - 972页]允许基于电子密度图的似然性来计算相位概率,该计算可独立于任何先验相位概率的计算进行。在此表明,仅从图似然函数计算得到的相位概率分布可能非常准确,并且它们对用于启动计算的相位显示出最小的偏差。图似然相位概率取决于电子密度图的预期特征,例如定义的溶剂区域以及溶剂区域和大分子占据区域内预期的电子密度分布。在最简单的情况下,图似然相位概率分布主要基于溶剂区域的平坦度。尽管图似然相位可以在没有先验相位信息的情况下计算,但高质量的起始相位会极大地增强它们。这导致了用于消除模型偏差的“初始化并切换”相位技术。在“初始化并切换”相位技术中,诸如来自模型的有偏差相位被用于初始化或启动图似然相位计算,然后最终相位仅从图似然相位计算中获得。图似然相位计算可应用于溶剂含量低至30%的情况。图似然相位计算的潜在应用包括从分子置换模型进行无偏差相位计算、迭代模型构建、在当前会使用2F(o) - F(c)或σ(A)加权图的情况下生成无偏差电子密度图、结构验证以及从溶剂掩码、非晶体学对称性或其他关于预期电子密度的知识进行从头相位确定。