Haas Brian J, Volfovsky Natalia, Town Christopher D, Troukhan Maxim, Alexandrov Nickolai, Feldmann Kenneth A, Flavell Richard B, White Owen, Salzberg Steven L
The Institute for Genomic Research, 9712 Medical Center Drive, Rockville, MD 20850, USA.
Genome Biol. 2002;3(6):RESEARCH0029. doi: 10.1186/gb-2002-3-6-research0029. Epub 2002 May 30.
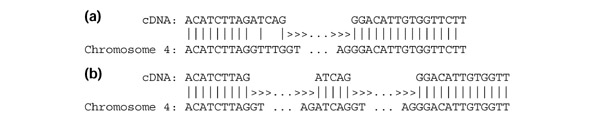
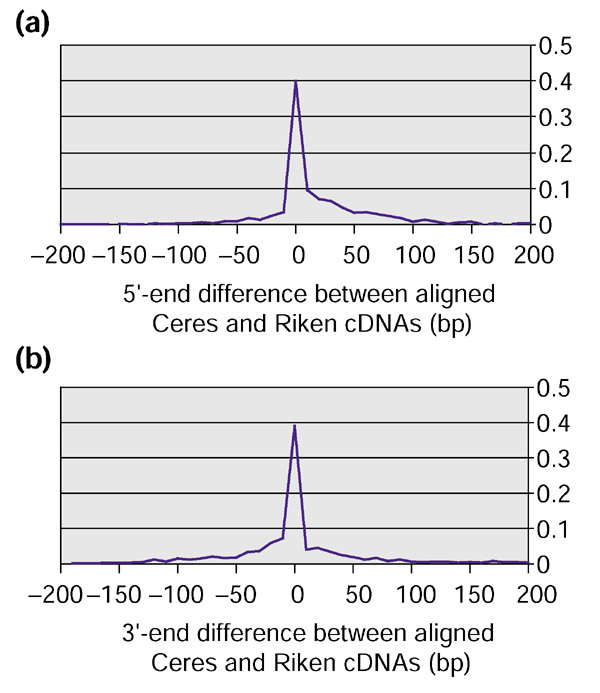
Annotation of eukaryotic genomes is a complex endeavor that requires the integration of evidence from multiple, often contradictory, sources. With the ever-increasing amount of genome sequence data now available, methods for accurate identification of large numbers of genes have become urgently needed. In an effort to create a set of very high-quality gene models, we used the sequence of 5,000 full-length gene transcripts from Arabidopsis to re-annotate its genome. We have mapped these transcripts to their exact chromosomal locations and, using alignment programs, have created gene models that provide a reference set for this organism.
Approximately 35% of the transcripts indicated that previously annotated genes needed modification, and 5% of the transcripts represented newly discovered genes. We also discovered that multiple transcription initiation sites appear to be much more common than previously known, and we report numerous cases of alternative mRNA splicing. We include a comparison of different alignment software and an analysis of how the transcript data improved the previously published annotation.
Our results demonstrate that sequencing of large numbers of full-length transcripts followed by computational mapping greatly improves identification of the complete exon structures of eukaryotic genes. In addition, we are able to find numerous introns in the untranslated regions of the genes.
真核生物基因组注释是一项复杂的工作,需要整合来自多个常常相互矛盾的数据源的证据。随着现在可用的基因组序列数据量不断增加,准确识别大量基因的方法变得迫切需要。为了创建一组非常高质量的基因模型,我们使用了来自拟南芥的5000个全长基因转录本的序列来重新注释其基因组。我们已将这些转录本定位到它们的确切染色体位置,并使用比对程序创建了为该生物体提供参考集的基因模型。
大约35%的转录本表明先前注释的基因需要修改,5%的转录本代表新发现的基因。我们还发现多个转录起始位点似乎比以前所知的更为常见,并且我们报告了许多可变mRNA剪接的情况。我们包括了不同比对软件的比较以及转录本数据如何改进先前发表的注释的分析。
我们的结果表明,大量全长转录本测序后进行计算定位极大地改善了真核生物基因完整外显子结构的识别。此外,我们能够在基因的非翻译区域发现大量内含子。