Schein Catherine H, Ozgün Numan, Izumi Tadahide, Braun Werner
Sealy Center for Structural Biology, Department of Human Biological Chemistry and Genetics, University of Texas Medical Branch, Galveston TX 77555-1157, USA.
BMC Bioinformatics. 2002 Nov 25;3:37. doi: 10.1186/1471-2105-3-37.
Total sequence decomposition, using the web-based MASIA tool, identifies areas of conservation in aligned protein sequences. By structurally annotating these motifs, the sequence can be parsed into individual building blocks, molecular legos ("molegos"), that can eventually be related to function. Here, the approach is applied to the apurinic/apyrimidinic endonuclease (APE) DNA repair proteins, essential enzymes that have been highly conserved throughout evolution. The APEs, DNase-1 and inositol 5'-polyphosphate phosphatases (IPP) form a superfamily that catalyze metal ion based phosphorolysis, but recognize different substrates.
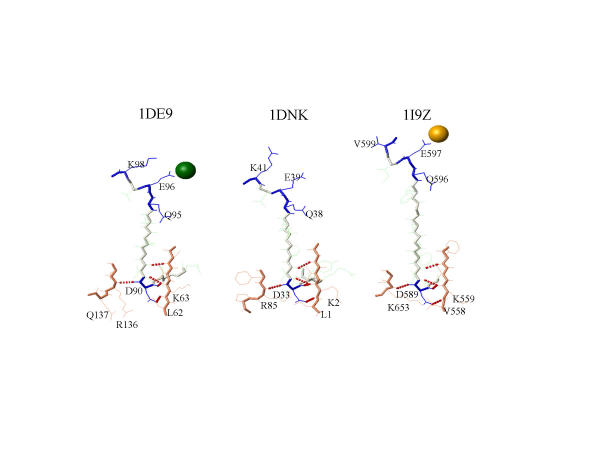
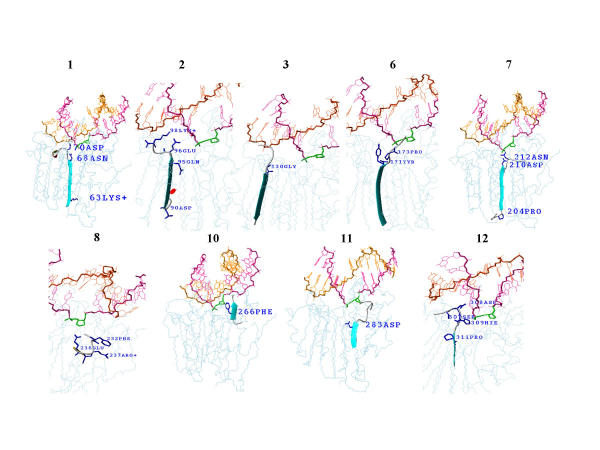
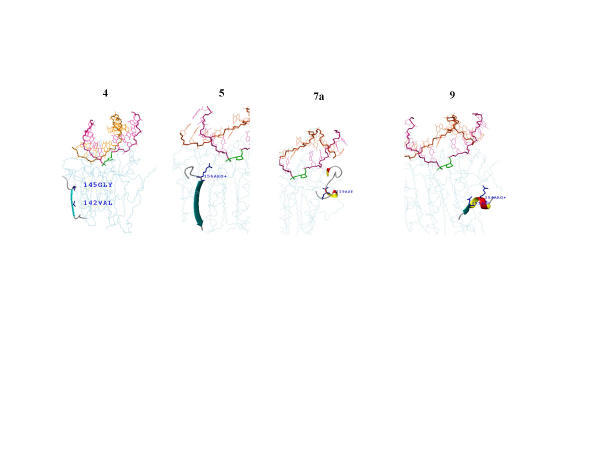
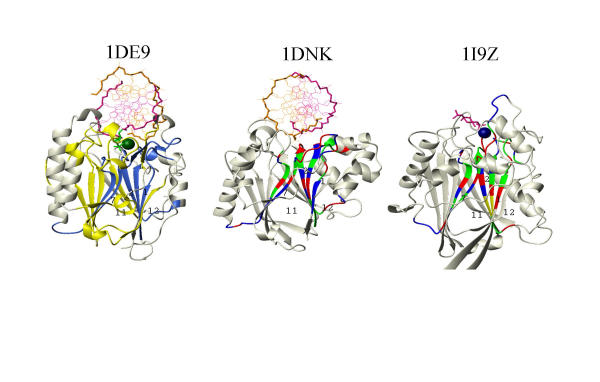
MASIA decomposition of APE yielded 12 sequence motifs, 10 of which are also structurally conserved within the family and are designated as molegos. The 12 motifs include all the residues known to be essential for DNA cleavage by APE. Five of these molegos are sequentially and structurally conserved in DNase-1 and the IPP family. Correcting the sequence alignment to match the residues at the ends of two of the molegos that are absolutely conserved in each of the three families greatly improved the local structural alignment of APEs, DNase-1 and synaptojanin. Comparing substrate/product binding of molegos common to DNase-1 showed that those distinctive for APEs are not directly involved in cleavage, but establish protein-DNA interactions 3' to the abasic site. These additional bonds enhance both specific binding to damaged DNA and the processivity of APE1.
A modular approach can improve structurally predictive alignments of homologous proteins with low sequence identity and reveal residues peripheral to the traditional "active site" that control the specificity of enzymatic activity.
使用基于网络的MASIA工具进行全序列分解,可识别比对后的蛋白质序列中的保守区域。通过对这些基序进行结构注释,序列可被解析为单个构建模块,即分子乐高积木(“分子乐高”),最终可将其与功能联系起来。在此,该方法应用于脱嘌呤/脱嘧啶内切核酸酶(APE)DNA修复蛋白,这是一类在整个进化过程中高度保守的必需酶。APE、DNase-1和肌醇5'-多磷酸磷酸酶(IPP)形成一个超家族,它们催化基于金属离子的磷酸解作用,但识别不同的底物。
APE的MASIA分解产生了12个序列基序,其中10个在该家族中也具有结构保守性,并被指定为分子乐高。这12个基序包括所有已知对APE切割DNA至关重要的残基。其中5个分子乐高在DNase-1和IPP家族中具有序列和结构保守性。校正序列比对以匹配在三个家族中每个家族都绝对保守的两个分子乐高末端的残基,极大地改善了APE、DNase-1和突触素的局部结构比对。比较DNase-1共有的分子乐高的底物/产物结合情况表明,APE特有的那些分子乐高并不直接参与切割,而是在无碱基位点的3'端建立蛋白质-DNA相互作用。这些额外的键增强了对受损DNA的特异性结合以及APE1的持续性。
模块化方法可以改善低序列同一性同源蛋白的结构预测比对,并揭示传统“活性位点”外围控制酶活性特异性的残基。