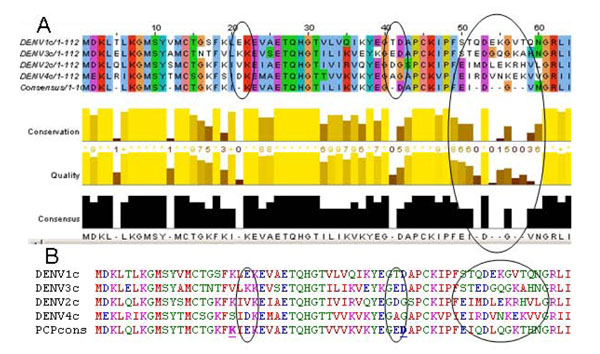
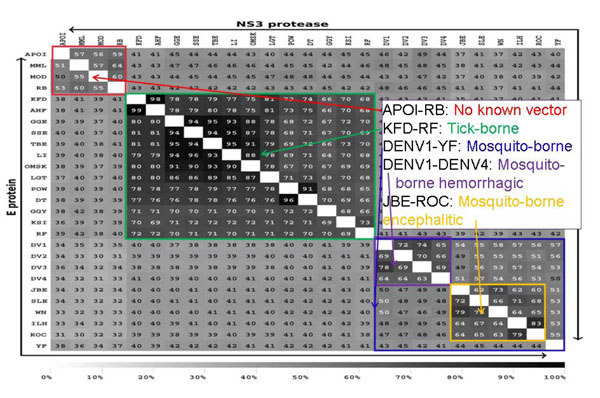
Institute for Translational Sciences, Computational Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Texas 77555-0857, USA.
BMC Bioinformatics. 2012;13 Suppl 13(Suppl 13):S9. doi: 10.1186/1471-2105-13-S13-S9. Epub 2012 Aug 24.
Analysis of large sets of biological sequence data from related strains or organisms is complicated by superficial redundancy in the set, which may contain many members that are identical except at one or two positions. Thus a new method, based on deriving physicochemical property (PCP)-consensus sequences, was tested for its ability to generate reference sequences and distinguish functionally significant changes from background variability.
The PCP consensus program was used to automatically derive consensus sequences starting from sequence alignments of proteins from Flaviviruses (from the Flavitrack database) and human enteroviruses, using a five dimensional set of Eigenvectors that summarize over 200 different scalar values for the PCPs of the amino acids. A PCP-consensus protein of a Dengue virus envelope protein was produced recombinantly and tested for its ability to bind antibodies to strains using ELISA.
PCP-consensus sequences of the flavivirus family could be used to classify them into five discrete groups and distinguish areas of the envelope proteins that correlate with host specificity and disease type. A multivalent Dengue virus antigen was designed and shown to bind antibodies against all four DENV types. A consensus enteroviral VPg protein had the same distinctive high pKa as wild type proteins and was recognized by two different polymerases.
The process for deriving PCP-consensus sequences for any group of aligned similar sequences, has been validated for sequences with up to 50% diversity. Ongoing projects have shown that the method identifies residues that significantly alter PCPs at a given position, and might thus cause changes in function or immunogenicity. Other potential applications include deriving target proteins for drug design and diagnostic kits.
对来自相关菌株或生物体的大量生物序列数据进行分析很复杂,因为这些数据集中存在表面冗余,其中可能包含许多除一两个位置外完全相同的成员。因此,我们测试了一种新的方法,该方法基于推导物理化学特性(PCP)-一致序列,以确定其生成参考序列并区分功能重要变化与背景变异的能力。
使用 PCP 一致程序,从 Flaviviruses(来自 Flavitrack 数据库)和人类肠病毒的蛋白质序列比对出发,自动推导一致序列,使用五维特征向量集总结氨基酸的 200 多种不同标量值的 PCP。使用 ELISA 测试了从重组方式产生的登革热病毒包膜蛋白的 PCP 一致蛋白,以确定其结合针对不同株的抗体的能力。
可以使用黄病毒科的 PCP 一致序列将它们分为五个离散组,并区分与宿主特异性和疾病类型相关的包膜蛋白区域。设计了一种多价登革热病毒抗原,并证明它可以与所有四种 DENV 型结合抗体。一致的肠病毒 VPg 蛋白具有与野生型蛋白相同的独特高 pKa 值,并且被两种不同的聚合酶识别。
已经验证了用于推导任何一组对齐相似序列的 PCP 一致序列的过程,对于具有高达 50%多样性的序列有效。正在进行的项目表明,该方法可识别在给定位置显着改变 PCP 的残基,因此可能导致功能或免疫原性改变。其他潜在应用包括为药物设计和诊断试剂盒推导靶蛋白。