Vandenesch Francois, Naimi Timothy, Enright Mark C, Lina Gerard, Nimmo Graeme R, Heffernan Helen, Liassine Nadia, Bes Michèle, Greenland Timothy, Reverdy Marie-Elisabeth, Etienne Jerome
INSERM E0230, Lyon, France.
Emerg Infect Dis. 2003 Aug;9(8):978-84. doi: 10.3201/eid0908.030089.
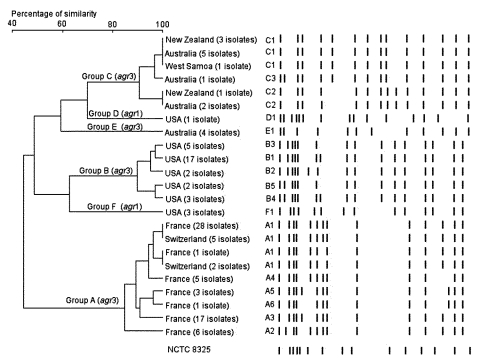
Infections caused by community-acquired (CA)-methicillin--resistant Staphylococcus aureus (MRSA) have been reported worldwide. We assessed whether any common genetic markers existed among 117 CA-MRSA isolates from the United States, France, Switzerland, Australia, New Zealand, and Western Samoa by performing polymerase chain reaction for 24 virulence factors and the methicillin-resistance determinant. The genetic background of the strain was analyzed by pulsed-field gel electrophoresis (PFGE) and multi-locus sequence typing (MLST). The CA-MRSA strains shared a type IV SCCmec cassette and the Panton-Valentine leukocidin locus, whereas the distribution of the other toxin genes was quite specific to the strains from each continent. PFGE and MLST analysis indicated distinct genetic backgrounds associated with each geographic origin, although predominantly restricted to the agr3 background. Within each continent, the genetic background of CA-MRSA strains did not correspond to that of the hospital-acquired MRSA.
社区获得性(CA)耐甲氧西林金黄色葡萄球菌(MRSA)引起的感染已在全球范围内报道。我们通过对24种毒力因子和甲氧西林耐药决定簇进行聚合酶链反应,评估了来自美国、法国、瑞士、澳大利亚、新西兰和西萨摩亚的117株CA-MRSA分离株中是否存在任何常见的遗传标记。通过脉冲场凝胶电泳(PFGE)和多位点序列分型(MLST)分析菌株的遗传背景。CA-MRSA菌株共享IV型SCCmec盒和杀白细胞素基因座,而其他毒素基因的分布在每个大陆的菌株中相当特异。PFGE和MLST分析表明,与每个地理来源相关的遗传背景不同,尽管主要限于agr3背景。在每个大陆内,CA-MRSA菌株的遗传背景与医院获得性MRSA的遗传背景不一致。