Nazina Anna G, Papatsenko Dmitri A
Department of Biology, New York University, New York, USA.
BMC Bioinformatics. 2003 Dec 22;4:65. doi: 10.1186/1471-2105-4-65.
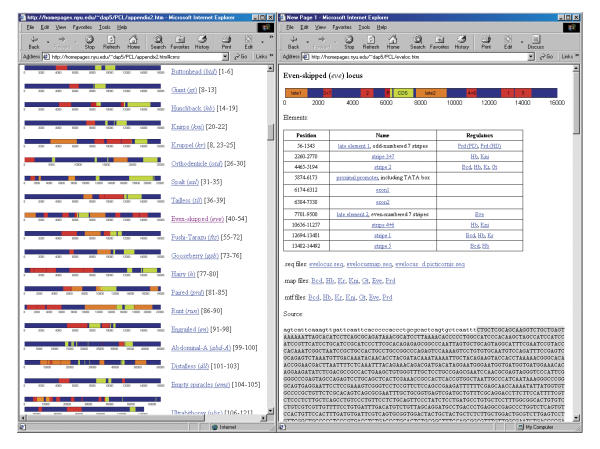
Transcription regulatory regions in higher eukaryotes are often represented by cis-regulatory modules (CRM) and are responsible for the formation of specific spatial and temporal gene expression patterns. These extended, approximately 1 KB, regions are found far from coding sequences and cannot be extracted from genome on the basis of their relative position to the coding regions.
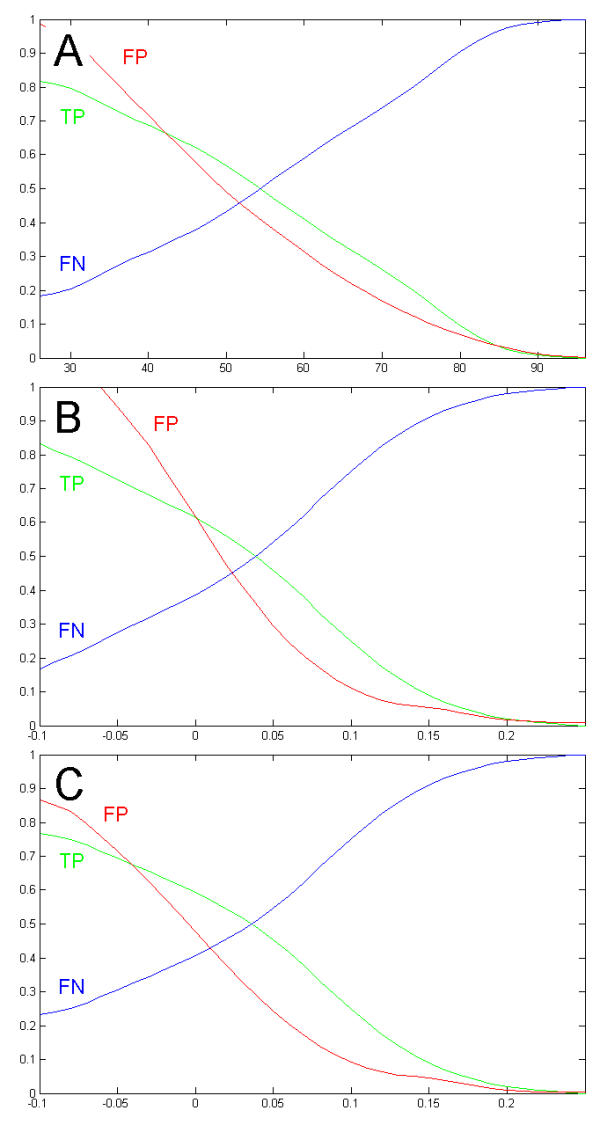
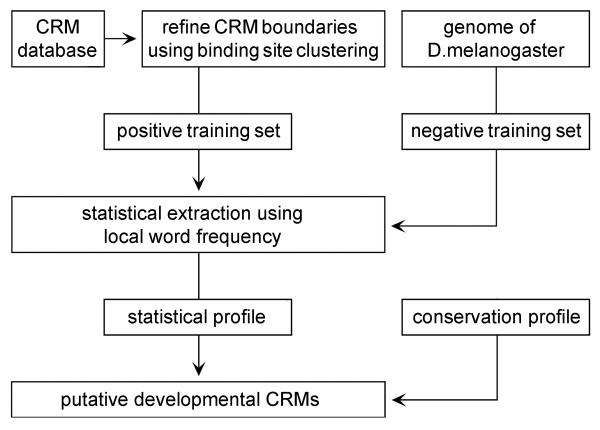
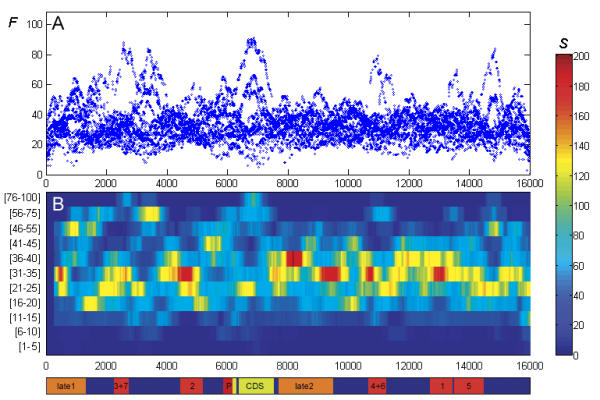
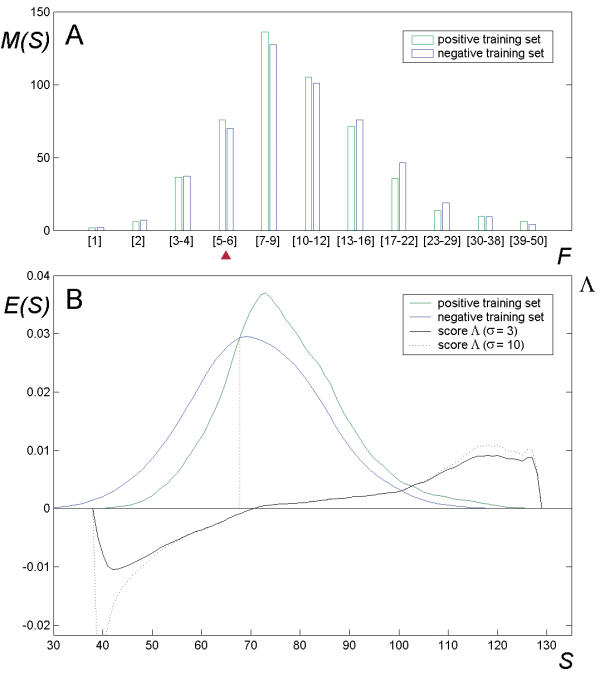
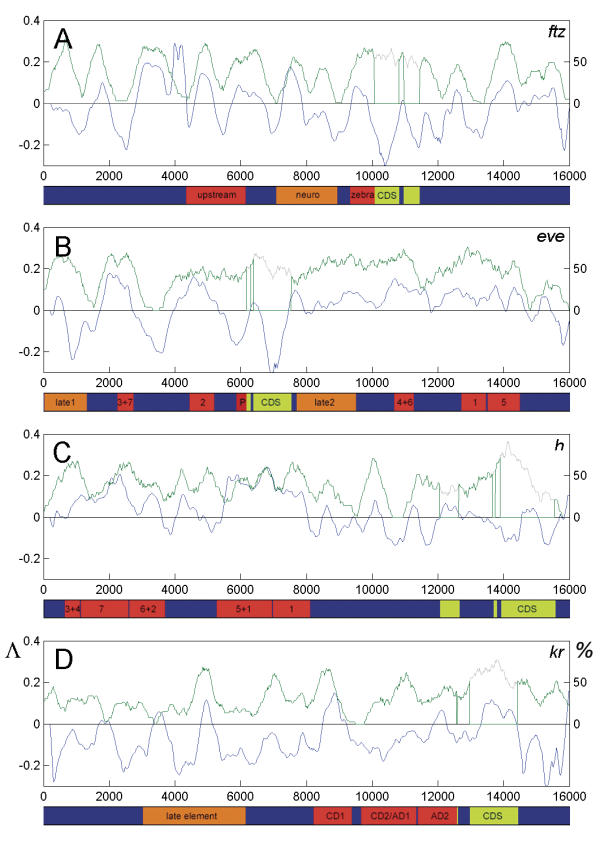
To explore the feasibility of CRM extraction from a genome, we generated an original training set, containing annotated sequence data for most of the known developmental CRMs from Drosophila. Based on this set of experimental data, we developed a strategy for statistical extraction of cis-regulatory modules from the genome, using exhaustive analysis of local word frequency (LWF). To assess the performance of our analysis, we measured the correlation between predictions generated by the LWF algorithm and the distribution of conserved non-coding regions in a number of Drosophila developmental genes.
In most of the cases tested, we observed high correlation (up to 0.6-0.8, measured on the entire gene locus) between the two independent techniques. We discuss computational strategies available for extraction of Drosophila CRMs and possible extensions of these methods.
高等真核生物中的转录调控区域通常由顺式调控模块(CRM)表示,并负责特定时空基因表达模式的形成。这些长度约为1千碱基对的扩展区域位于远离编码序列的位置,无法根据其与编码区域的相对位置从基因组中提取。
为了探索从基因组中提取CRM的可行性,我们生成了一个原始训练集,其中包含来自果蝇的大多数已知发育CRM的注释序列数据。基于这组实验数据,我们开发了一种利用局部词频(LWF)详尽分析从基因组中统计提取顺式调控模块的策略。为了评估我们分析的性能,我们测量了LWF算法生成的预测与一些果蝇发育基因中保守非编码区域分布之间的相关性。
在大多数测试案例中,我们观察到这两种独立技术之间具有高度相关性(在整个基因座上测量,高达0.6 - 0.8)。我们讨论了可用于提取果蝇CRM的计算策略以及这些方法可能的扩展。