Abba Martin C, Drake Jeffrey A, Hawkins Kathleen A, Hu Yuhui, Sun Hongxia, Notcovich Cintia, Gaddis Sally, Sahin Aysegul, Baggerly Keith, Aldaz C Marcelo
Department of Carcinogenesis, The University of Texas MD Anderson Cancer Center, Science Park - Research Division, Smithville, Texas, USA.
Breast Cancer Res. 2004;6(5):R499-513. doi: 10.1186/bcr899. Epub 2004 Jul 6.
Genomic and transcriptomic alterations affecting key cellular processes such us cell proliferation, differentiation and genomic stability are considered crucial for the development and progression of cancer. Most invasive breast carcinomas are known to derive from precursor in situ lesions. It is proposed that major global expression abnormalities occur in the transition from normal to premalignant stages and further progression to invasive stages. Serial analysis of gene expression (SAGE) was employed to generate a comprehensive global gene expression profile of the major changes occurring during breast cancer malignant evolution.
In the present study we combined various normal and tumor SAGE libraries available in the public domain with sets of breast cancer SAGE libraries recently generated and sequenced in our laboratory. A recently developed modified t test was used to detect the genes differentially expressed.
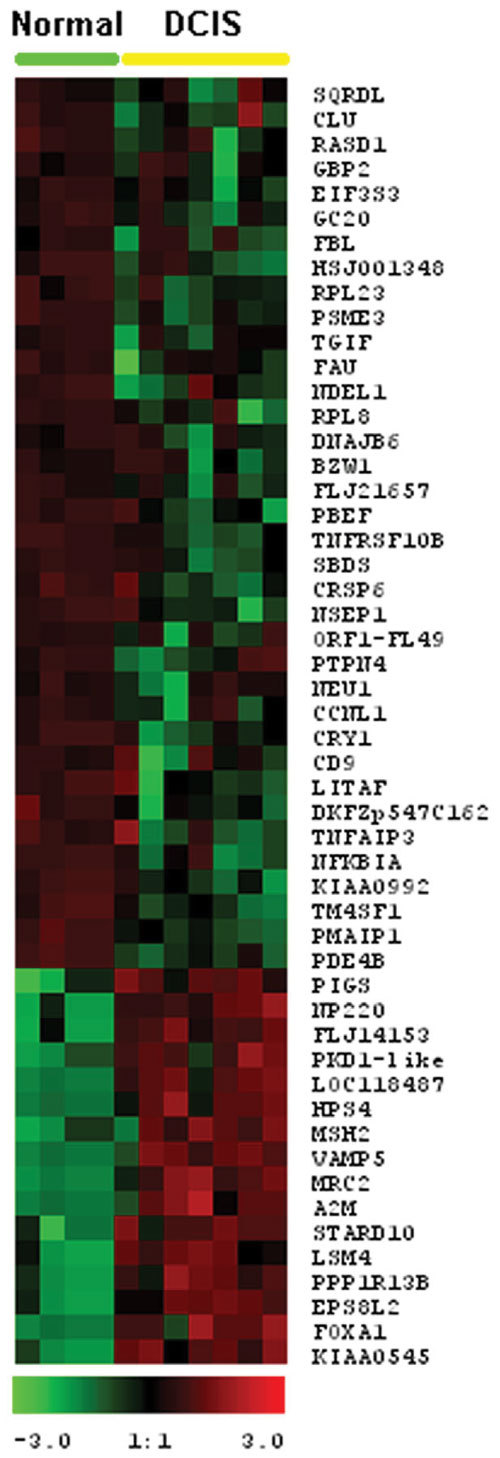
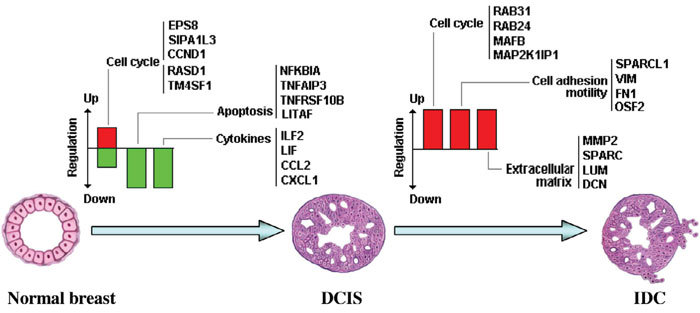
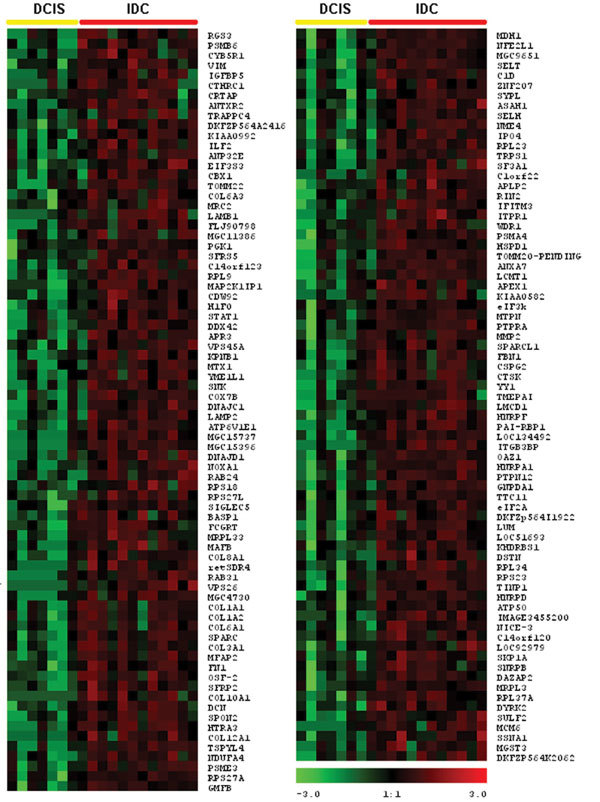
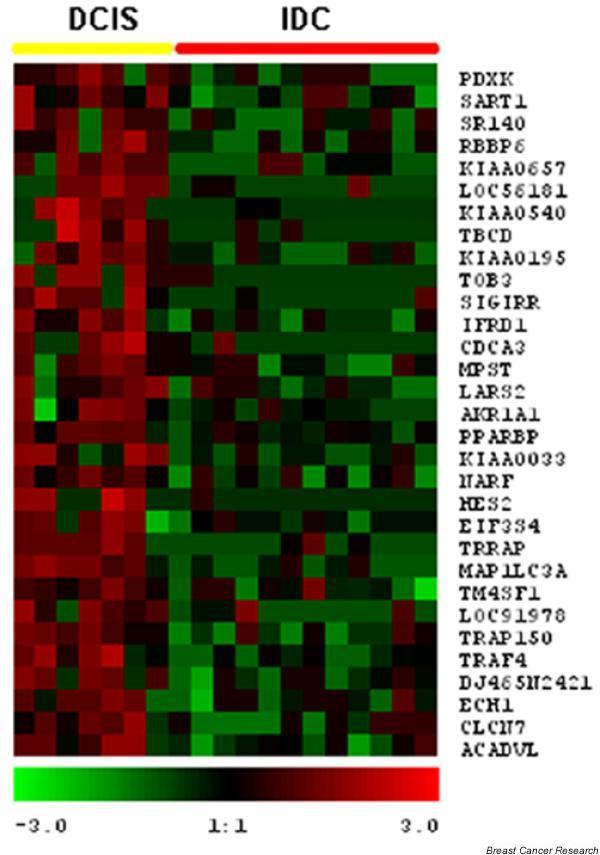
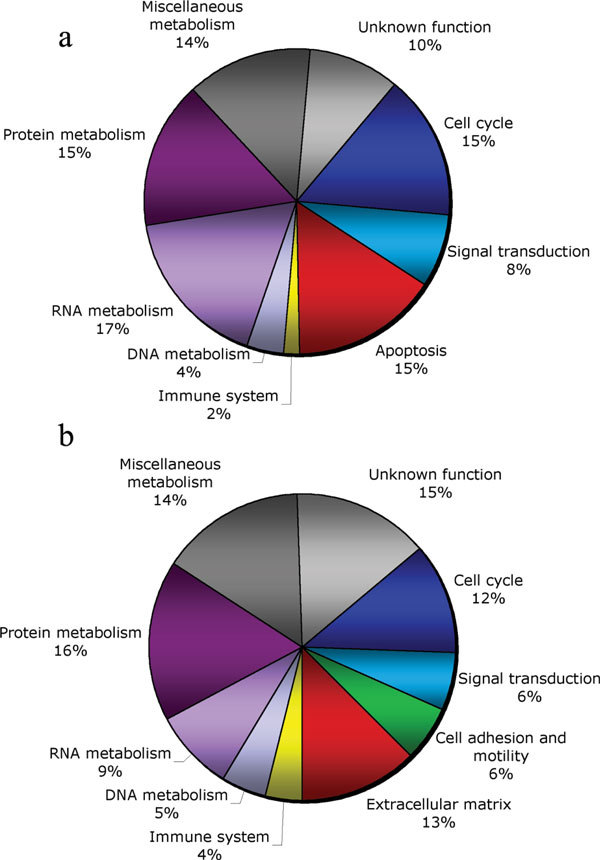
We accumulated a total of approximately 1.7 million breast tissue-specific SAGE tags and monitored the behavior of more than 25,157 genes during early breast carcinogenesis. We detected 52 transcripts commonly deregulated across the board when comparing normal tissue with ductal carcinoma in situ, and 149 transcripts when comparing ductal carcinoma in situ with invasive ductal carcinoma (P < 0.01).
A major novelty of our study was the use of a statistical method that correctly accounts for the intra-SAGE and inter-SAGE library sources of variation. The most useful result of applying this modified t statistics beta binomial test is the identification of genes and gene families commonly deregulated across samples within each specific stage in the transition from normal to preinvasive and invasive stages of breast cancer development. Most of the gene expression abnormalities detected at the in situ stage were related to specific genes in charge of regulating the proper homeostasis between cell death and cell proliferation. The comparison of in situ lesions with fully invasive lesions, a much more heterogeneous group, clearly identified as the most importantly deregulated group of transcripts those encoding for various families of proteins in charge of extracellular matrix remodeling, invasion and cell motility functions.
影响细胞增殖、分化和基因组稳定性等关键细胞过程的基因组和转录组改变被认为对癌症的发生和发展至关重要。大多数浸润性乳腺癌已知起源于原位前体病变。有人提出,在从正常阶段到癌前阶段以及进一步发展到浸润阶段的转变过程中会发生主要的全局表达异常。采用基因表达系列分析(SAGE)来生成乳腺癌恶性演变过程中发生的主要变化的全面全局基因表达谱。
在本研究中,我们将公共领域中可用的各种正常和肿瘤SAGE文库与我们实验室最近生成并测序的一组乳腺癌SAGE文库相结合。使用最近开发的改良t检验来检测差异表达的基因。
我们总共积累了约170万个乳腺组织特异性SAGE标签,并监测了早期乳腺癌发生过程中超过25157个基因的行为。在将正常组织与原位导管癌进行比较时,我们检测到52个转录本普遍失调,而在将原位导管癌与浸润性导管癌进行比较时,检测到149个转录本(P < 0.01)。
我们研究的一个主要新颖之处在于使用了一种统计方法,该方法能够正确考虑SAGE内部和SAGE文库间的变异来源。应用这种改良的t统计β二项式检验最有用的结果是鉴定出在乳腺癌从正常发展到侵袭前和侵袭阶段的每个特定阶段中,跨样本普遍失调的基因和基因家族。在原位阶段检测到的大多数基因表达异常与负责调节细胞死亡和细胞增殖之间适当稳态的特定基因有关。将原位病变与完全浸润性病变(一个更加异质的组)进行比较,清楚地确定编码负责细胞外基质重塑、侵袭和细胞运动功能的各种蛋白质家族的转录本是失调最严重的组。