Schlicht Michael, Matysiak Brian, Brodzeller Tracy, Wen Xinyu, Liu Hang, Zhou Guohui, Dhir Rajiv, Hessner Martin J, Tonellato Peter, Suckow Mark, Pollard Morris, Datta Milton W
Department of Pathology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI, 53226, USA.
BMC Genomics. 2004 Aug 20;5(1):58. doi: 10.1186/1471-2164-5-58.
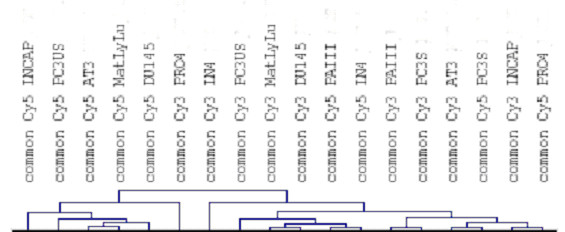
Gene expression technologies have the ability to generate vast amounts of data, yet there often resides only limited resources for subsequent validation studies. This necessitates the ability to perform sorting and prioritization of the output data. Previously described methodologies have used functional pathways or transcriptional regulatory grouping to sort genes for further study. In this paper we demonstrate a comparative genomics based method to leverage data from animal models to prioritize genes for validation. This approach allows one to develop a disease-based focus for the prioritization of gene data, a process that is essential for systems that lack significant functional pathway data yet have defined animal models. This method is made possible through the use of highly controlled spotted cDNA slide production and the use of comparative bioinformatics databases without the use of cross-species slide hybridizations.
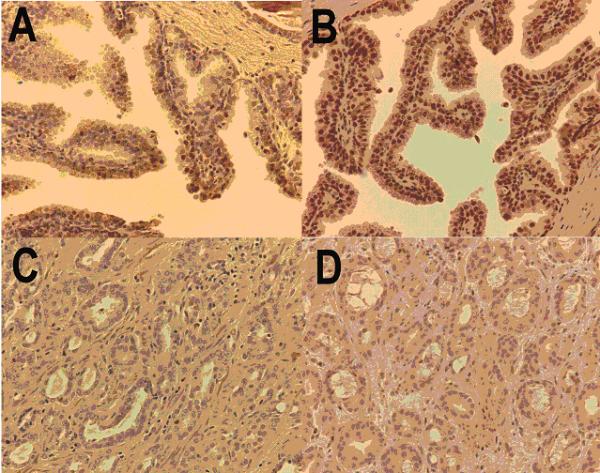
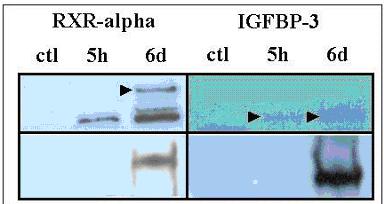
Using gene expression profiling we have demonstrated a similar whole transcriptome gene expression patterns in prostate cancer cells from human and rat prostate cancer cell lines both at baseline expression levels and after treatment with physiologic concentrations of the proposed chemopreventive agent Selenium. Using both the human PC3 and rat PAII prostate cancer cell lines have gone on to identify a subset of one hundred and fifty-four genes that demonstrate a similar level of differential expression to Selenium treatment in both species. Further analysis and data mining for two genes, the Insulin like Growth Factor Binding protein 3, and Retinoic X Receptor alpha, demonstrates an association with prostate cancer, functional pathway links, and protein-protein interactions that make these genes prime candidates for explaining the mechanism of Selenium's chemopreventive effect in prostate cancer. These genes are subsequently validated by western blots showing Selenium based induction and using tissue microarrays to demonstrate a significant association between downregulated protein expression and tumorigenesis, a process that is the reverse of what is seen in the presence of Selenium.
Thus the outlined process demonstrates similar baseline and selenium induced gene expression profiles between rat and human prostate cancers, and provides a method for identifying testable functional pathways for the action of Selenium's chemopreventive properties in prostate cancer.
基因表达技术能够生成海量数据,但后续验证研究的资源往往有限。这就需要具备对输出数据进行分类和排序的能力。先前描述的方法利用功能通路或转录调控分组来对基因进行分类以便进一步研究。在本文中,我们展示了一种基于比较基因组学的方法,利用动物模型的数据对基因进行排序以便验证。这种方法使人们能够针对基因数据的排序建立基于疾病的重点关注,对于那些缺乏大量功能通路数据但已定义动物模型的系统而言,这一过程至关重要。通过使用高度可控的点阵cDNA芯片制作以及比较生物信息学数据库,而不使用跨物种芯片杂交,使得该方法成为可能。
利用基因表达谱分析,我们证明了在基线表达水平以及用生理浓度的拟化学预防剂硒处理后,人前列腺癌细胞系和大鼠前列腺癌细胞系中的全转录组基因表达模式相似。使用人PC3和大鼠PAII前列腺癌细胞系,进而鉴定出了154个基因的一个子集,这些基因在两个物种中对硒处理均表现出相似水平的差异表达。对两个基因,即胰岛素样生长因子结合蛋白3和视黄酸X受体α进行进一步分析和数据挖掘,证明它们与前列腺癌、功能通路联系以及蛋白质 - 蛋白质相互作用相关,这使得这些基因成为解释硒在前列腺癌中化学预防作用机制的主要候选基因。随后通过蛋白质印迹法验证这些基因,显示出基于硒的诱导作用,并使用组织微阵列证明下调的蛋白质表达与肿瘤发生之间存在显著关联,这一过程与在有硒存在时所观察到的情况相反。
因此,所概述的过程证明了大鼠和人前列腺癌之间相似的基线和硒诱导基因表达谱,并提供了一种方法来识别硒在前列腺癌中化学预防特性作用的可测试功能通路。