Fairbrother William G, Holste Dirk, Burge Christopher B, Sharp Phillip A
Center for Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA.
PLoS Biol. 2004 Sep;2(9):E268. doi: 10.1371/journal.pbio.0020268. Epub 2004 Aug 31.
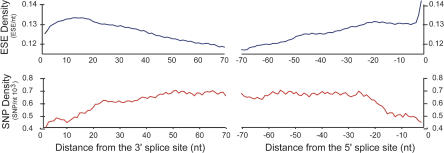
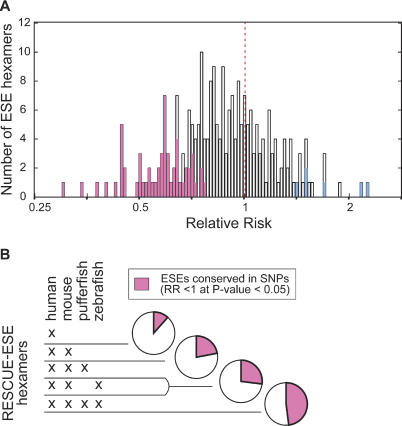
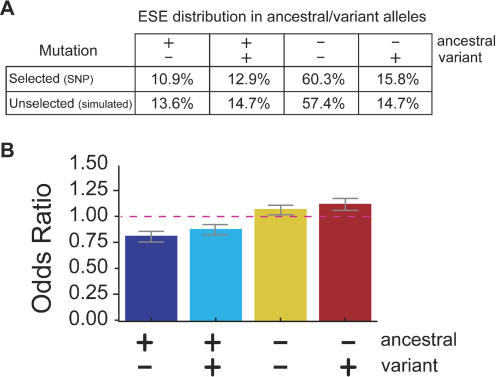
Because deleterious alleles arising from mutation are filtered by natural selection, mutations that create such alleles will be underrepresented in the set of common genetic variation existing in a population at any given time. Here, we describe an approach based on this idea called VERIFY (variant elimination reinforces functionality), which can be used to assess the extent of natural selection acting on an oligonucleotide motif or set of motifs predicted to have biological activity. As an application of this approach, we analyzed a set of 238 hexanucleotides previously predicted to have exonic splicing enhancer (ESE) activity in human exons using the relative enhancer and silencer classification by unanimous enrichment (RESCUE)-ESE method. Aligning the single nucleotide polymorphisms (SNPs) from the public human SNP database to the chimpanzee genome allowed inference of the direction of the mutations that created present-day SNPs. Analyzing the set of SNPs that overlap RESCUE-ESE hexamers, we conclude that nearly one-fifth of the mutations that disrupt predicted ESEs have been eliminated by natural selection (odds ratio = 0.82 +/- 0.05). This selection is strongest for the predicted ESEs that are located near splice sites. Our results demonstrate a novel approach for quantifying the extent of natural selection acting on candidate functional motifs and also suggest certain features of mutations/SNPs, such as proximity to the splice site and disruption or alteration of predicted ESEs, that should be useful in identifying variants that might cause a biological phenotype.
由于由突变产生的有害等位基因会受到自然选择的筛选,所以在任何给定时间,产生此类等位基因的突变在群体中存在的常见遗传变异集合中所占比例会较低。在此,我们描述了一种基于此理念的方法,称为VERIFY(变异消除强化功能性),该方法可用于评估自然选择对预测具有生物活性的寡核苷酸基序或一组基序的作用程度。作为该方法的一个应用,我们使用一致富集相对增强子和沉默子分类法(RESCUE)-ESE方法,分析了一组先前预测在人类外显子中具有外显子剪接增强子(ESE)活性的238个六核苷酸。将来自公共人类单核苷酸多态性(SNP)数据库的SNP与黑猩猩基因组进行比对,能够推断出产生当今SNP的突变方向。通过分析与RESCUE-ESE六聚体重叠的SNP集合,我们得出结论,破坏预测ESE的突变中近五分之一已被自然选择消除(优势比 = 0.82 ± 0.05)。这种选择对于位于剪接位点附近的预测ESE最为强烈。我们的结果展示了一种量化自然选择对候选功能基序作用程度的新方法,同时也揭示了突变/SNP的某些特征,如与剪接位点的距离以及对预测ESE的破坏或改变,这些特征在识别可能导致生物学表型的变异方面应该会很有用。