Storey John D, Akey Joshua M, Kruglyak Leonid
Department of Biostatistics, University of Washington, Seattle, Washington, USA.
PLoS Biol. 2005 Aug;3(8):e267. doi: 10.1371/journal.pbio.0030267. Epub 2005 Jul 26.
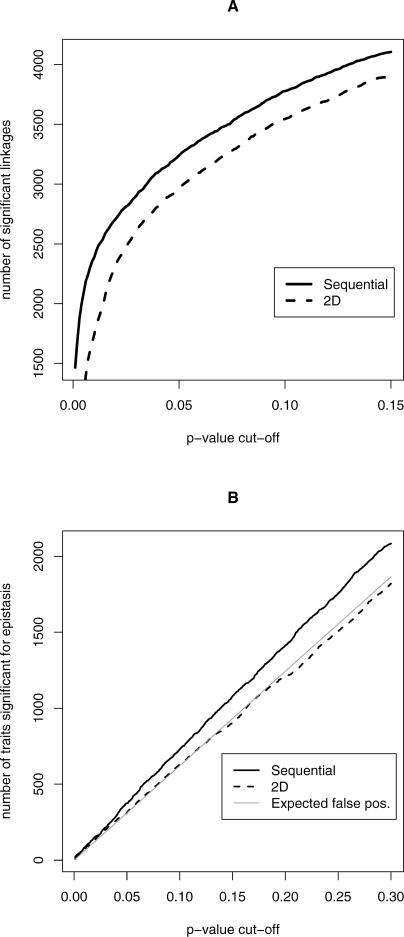
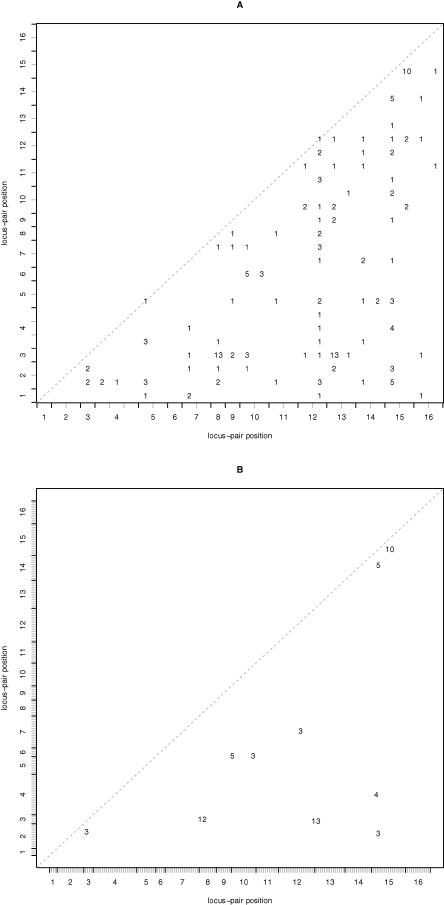
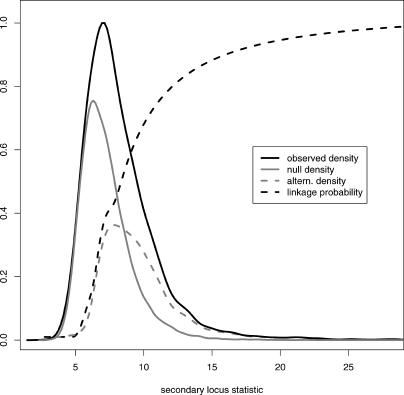
With the ability to measure thousands of related phenotypes from a single biological sample, it is now feasible to genetically dissect systems-level biological phenomena. The genetics of transcriptional regulation and protein abundance are likely to be complex, meaning that genetic variation at multiple loci will influence these phenotypes. Several recent studies have investigated the role of genetic variation in transcription by applying traditional linkage analysis methods to genomewide expression data, where each gene expression level was treated as a quantitative trait and analyzed separately from one another. Here, we develop a new, computationally efficient method for simultaneously mapping multiple gene expression quantitative trait loci that directly uses all of the available data. Information shared across gene expression traits is captured in a way that makes minimal assumptions about the statistical properties of the data. The method produces easy-to-interpret measures of statistical significance for both individual loci and the overall joint significance of multiple loci selected for a given expression trait. We apply the new method to a cross between two strains of the budding yeast Saccharomyces cerevisiae, and estimate that at least 37% of all gene expression traits show two simultaneous linkages, where we have allowed for epistatic interactions. Pairs of jointly linking quantitative trait loci are identified with high confidence for 170 gene expression traits, where it is expected that both loci are true positives for at least 153 traits. In addition, we are able to show that epistatic interactions contribute to gene expression variation for at least 14% of all traits. We compare the proposed approach to an exhaustive two-dimensional scan over all pairs of loci. Surprisingly, we demonstrate that an exhaustive two-dimensional scan is less powerful than the sequential search used here. In addition, we show that a two-dimensional scan does not truly allow one to test for simultaneous linkage, and the statistical significance measured from this existing method cannot be interpreted among many traits.
由于能够从单个生物样本中测量数千种相关表型,现在从遗传学角度剖析系统水平的生物学现象已切实可行。转录调控和蛋白质丰度的遗传学可能很复杂,这意味着多个基因座的遗传变异会影响这些表型。最近的几项研究通过将传统连锁分析方法应用于全基因组表达数据来研究遗传变异在转录中的作用,其中每个基因表达水平都被视为一个数量性状并相互独立分析。在此,我们开发了一种新的、计算效率高的方法,用于同时定位多个基因表达数量性状基因座,该方法直接使用所有可用数据。以对数据统计特性做出最小假设的方式捕获跨基因表达性状共享的信息。该方法为单个基因座以及为给定表达性状选择的多个基因座的整体联合显著性生成易于解释的统计显著性度量。我们将新方法应用于酿酒酵母两个菌株之间的杂交,并估计所有基因表达性状中至少37%显示出两个同时的连锁关系,其中我们考虑了上位性相互作用。对于170个基因表达性状,以高置信度鉴定出成对的联合连锁数量性状基因座,预计其中至少153个性状的两个基因座均为真阳性。此外,我们能够证明上位性相互作用至少对所有性状的14%的基因表达变异有贡献。我们将所提出的方法与对所有基因座对进行详尽的二维扫描进行比较。令人惊讶的是,我们证明详尽的二维扫描不如这里使用的顺序搜索有效。此外,我们表明二维扫描并不能真正允许人们测试同时连锁,并且从这种现有方法测量的统计显著性在许多性状之间无法解释。