Li Tian-Fang, Darowish Michael, Zuscik Michael J, Chen Di, Schwarz Edward M, Rosier Randy N, Drissi Hicham, O'Keefe Regis J
Center for Musculoskeletal Research, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA.
J Bone Miner Res. 2006 Jan;21(1):4-16. doi: 10.1359/JBMR.050911. Epub 2005 Sep 19.
Smad3 deficiency accelerates chondrocyte maturation and leads to osteoarthritis. Primary chondrocytes without Smad3 lack compensatory increases of TGF-beta signaling factors, but BMP-related gene expression is increased. Smad2 or Smad3 overexpression and BMP blockade abrogate accelerated maturation in Smad3-/- chondrocytes. BMP signaling is increased in TGF-beta deficiency and is required for accelerated chondrocyte maturation.
Disruption of TGF-beta signaling results in accelerated chondrocyte maturation and leads to postnatal dwarfism and premature osteoarthritis. The mechanisms involved in this process were studied using in vitro murine chondrocyte cultures.
Primary chondrocytes were isolated from the sterna of neonatal wildtype and Smad3-/- mice. Expressions of maturational markers, as well as genes involved in TGF-beta and BMP signaling were examined. Chondrocytes were treated with TGF-beta and BMP-2, and effects on maturation-related genes and BMP/TGF-beta responsive reporters were examined. Recombinant noggin or retroviral vectors expressing Smad2 or Smad3 were added to the cultures.
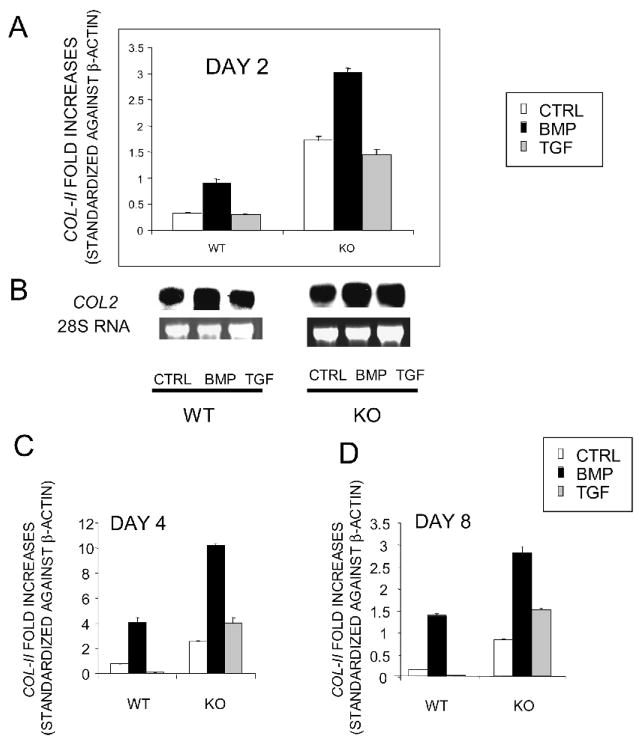
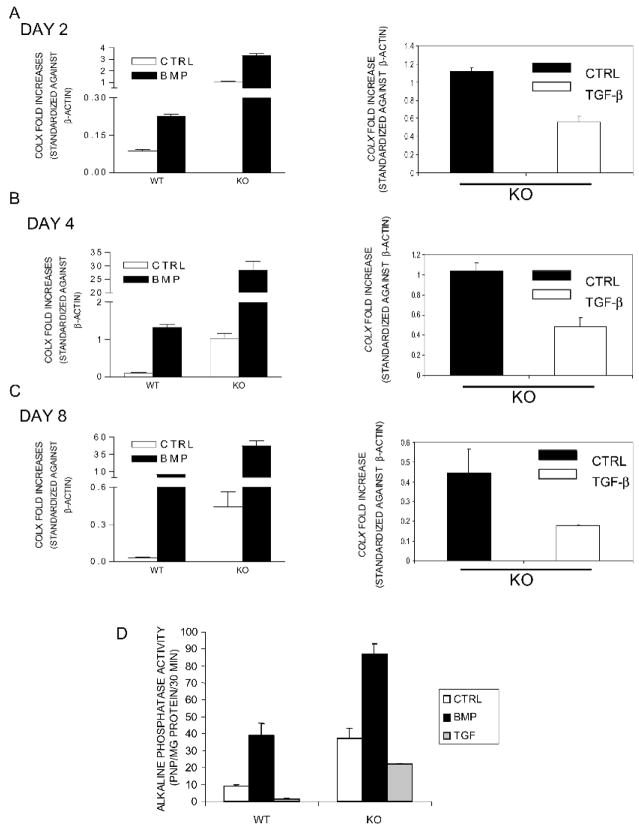
Expression of colX and other maturational markers was markedly increased in Smad3-/- chondrocytes. Smad3-/- chondrocytes lacked compensatory increases in Smad2, Smad4, TGFRII, Sno, or Smurf2 and had reduced expression of TGF-beta1 and TGFRI. In contrast, Smad1, Smad5, BMP2, and BMP6 expression was increased, suggesting a shift from TGF-beta toward BMP signaling. In Smad3-/- chondrocytes, alternative TGF-beta signaling pathways remained responsive, as shown by luciferase assays. These non-Smad3-dependent TGF-beta pathways reduced colX expression and alkaline phosphatase activity in TGF-beta-treated Smad3-/- cultures, but only partially. In contrast, Smad3-/- chondrocytes were more responsive to BMP-2 treatment and had increased colX expression, phosphoSmads 1, 5, and 8 levels, and luciferase reporter activity. Overexpression of both Smad2 and Smad3 blocked spontaneous maturation in Smad3-deficient chondrocytes. Maturation was also abrogated by the addition of noggin, an extracellular BMP inhibitor.
These findings show a key role for BMP signaling during the chondrocyte maturation, occurring with loss of TGF-beta signaling with important implications for osteoarthritis and cartilage diseases.
Smad3缺乏会加速软骨细胞成熟并导致骨关节炎。缺乏Smad3的原代软骨细胞缺乏TGF-β信号因子的代偿性增加,但BMP相关基因表达增加。Smad2或Smad3过表达以及BMP阻断可消除Smad3-/-软骨细胞的加速成熟。在TGF-β缺乏时BMP信号增强,且是软骨细胞加速成熟所必需的。
TGF-β信号的破坏导致软骨细胞加速成熟,并导致出生后侏儒症和早发性骨关节炎。使用体外小鼠软骨细胞培养物研究了该过程中涉及的机制。
从新生野生型和Smad3-/-小鼠的胸骨中分离原代软骨细胞。检测成熟标志物以及参与TGF-β和BMP信号的基因的表达。用TGF-β和BMP-2处理软骨细胞,并检测对成熟相关基因和BMP/TGF-β反应性报告基因的影响。将重组头蛋白或表达Smad2或Smad3的逆转录病毒载体添加到培养物中。
Smad3-/-软骨细胞中colX和其他成熟标志物的表达明显增加。Smad3-/-软骨细胞缺乏Smad2、Smad4、TGFRII、Sno或Smurf2的代偿性增加,并且TGF-β1和TGFRI的表达降低。相反,Smad1、Smad5、BMP2和BMP6的表达增加,表明从TGF-β信号向BMP信号转变。如荧光素酶测定所示,在Smad3-/-软骨细胞中,替代性TGF-β信号通路仍然有反应。这些非Smad3依赖性TGF-β通路降低了TGF-β处理的Smad3-/-培养物中的colX表达和碱性磷酸酶活性,但只是部分降低。相反,Smad3-/-软骨细胞对BMP-2处理更敏感,并且colX表达增加、磷酸化Smads 1、5和8水平以及荧光素酶报告基因活性增加。Smad2和Smad3的过表达阻断了Smad3缺陷软骨细胞的自发成熟。添加头蛋白(一种细胞外BMP抑制剂)也可消除成熟。
这些发现表明BMP信号在软骨细胞成熟过程中起关键作用,这发生在TGF-β信号丧失时,对骨关节炎和软骨疾病具有重要意义。