Tekaia Fredj, Yeramian Edouard
Unité de Génétique Moléculaire des Levures (URA 2171 CNRS and UFR927 Univ. P.M. Curie), Institut Pasteur, Paris, France.
PLoS Comput Biol. 2005 Dec;1(7):e75. doi: 10.1371/journal.pcbi.0010075. Epub 2005 Dec 16.
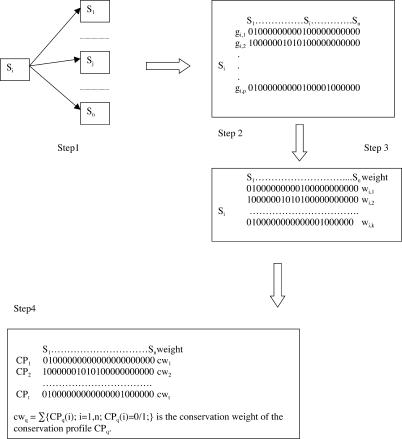
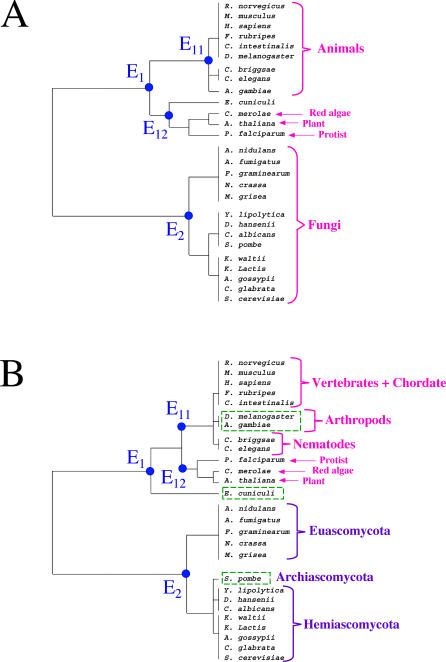
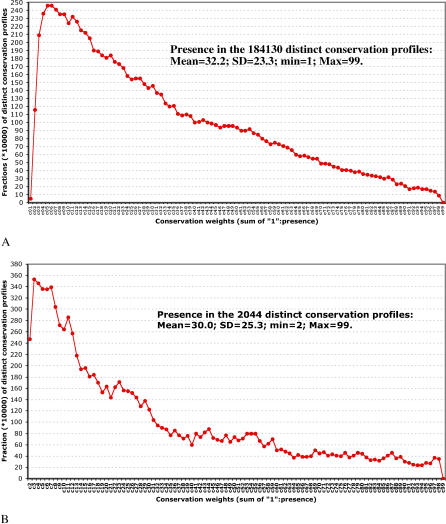
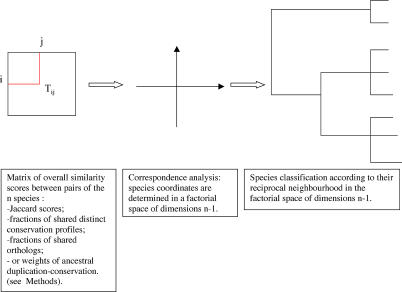
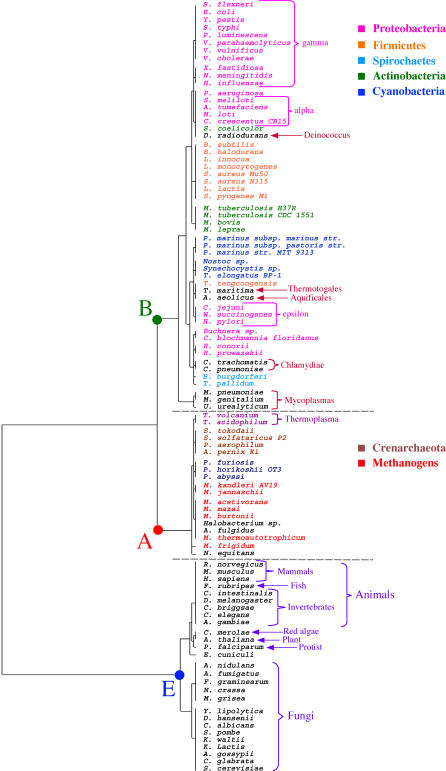
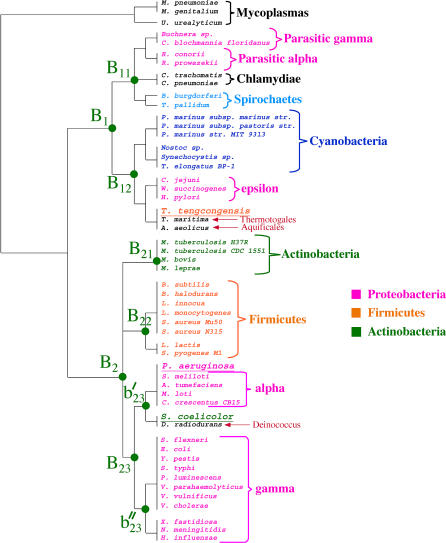
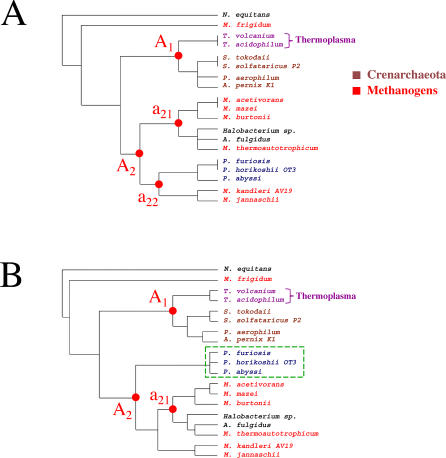
The concept of the genome tree depends on the potential evolutionary significance in the clustering of species according to similarities in the gene content of their genomes. In this respect, genome trees have often been identified with species trees. With the rapid expansion of genome sequence data it becomes of increasing importance to develop accurate methods for grasping global trends for the phylogenetic signals that mutually link the various genomes. We therefore derive here the methodological concept of genome trees based on protein conservation profiles in multiple species. The basic idea in this derivation is that the multi-component "presence-absence" protein conservation profiles permit tracking of common evolutionary histories of genes across multiple genomes. We show that a significant reduction in informational redundancy is achieved by considering only the subset of distinct conservation profiles. Beyond these basic ideas, we point out various pitfalls and limitations associated with the data handling, paving the way for further improvements. As an illustration for the methods, we analyze a genome tree based on the above principles, along with a series of other trees derived from the same data and based on pair-wise comparisons (ancestral duplication-conservation and shared orthologs). In all trees we observe a sharp discrimination between the three primary domains of life: Bacteria, Archaea, and Eukarya. The new genome tree, based on conservation profiles, displays a significant correspondence with classically recognized taxonomical groupings, along with a series of departures from such conventional clusterings.
基因组树的概念取决于根据物种基因组中基因含量的相似性对物种进行聚类的潜在进化意义。在这方面,基因组树常常与物种树等同起来。随着基因组序列数据的迅速扩展,开发准确的方法以把握相互关联的各种基因组的系统发育信号的全球趋势变得越来越重要。因此,我们在此基于多个物种的蛋白质保守概况推导基因组树的方法概念。这种推导的基本思想是,多组分的“存在 - 缺失”蛋白质保守概况允许追踪多个基因组中基因的共同进化历史。我们表明,仅考虑不同保守概况的子集就能显著减少信息冗余。除了这些基本思想,我们指出了与数据处理相关的各种陷阱和局限性,为进一步改进铺平道路。作为该方法的一个示例,我们基于上述原则分析了一个基因组树,以及一系列从相同数据并基于成对比较(祖先复制 - 保守和共享直系同源物)得出的其他树。在所有的树中,我们都观察到生命的三个主要域(细菌、古菌和真核生物)之间有明显的区分。基于保守概况的新基因组树与经典认可的分类分组有显著的对应关系,同时也与这些传统聚类有一系列的偏离。