Huang Jing, Wei Wen, Chen Joyce, Zhang Jane, Liu Guoying, Di Xiaojun, Mei Rui, Ishikawa Shumpei, Aburatani Hiroyuki, Jones Keith W, Shapero Michael H
Affymetrix, Inc, 3420 Central Expressway, Santa Clara, CA 95051, USA.
BMC Bioinformatics. 2006 Feb 21;7:83. doi: 10.1186/1471-2105-7-83.
DNA copy number alterations are one of the main characteristics of the cancer cell karyotype and can contribute to the complex phenotype of these cells. These alterations can lead to gains in cellular oncogenes as well as losses in tumor suppressor genes and can span small intervals as well as involve entire chromosomes. The ability to accurately detect these changes is central to understanding how they impact the biology of the cell.
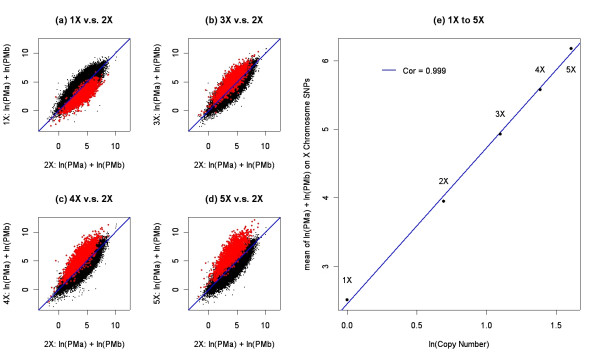
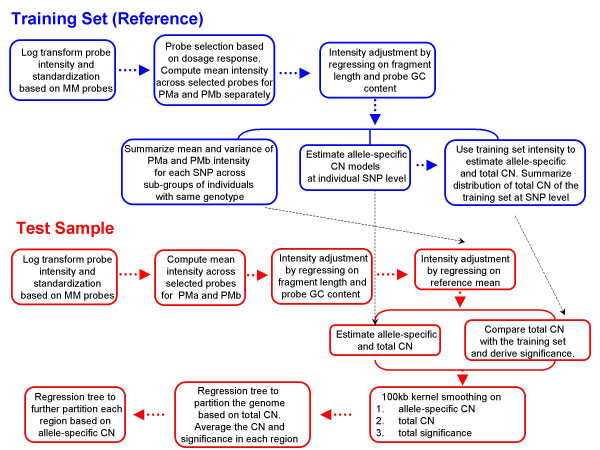
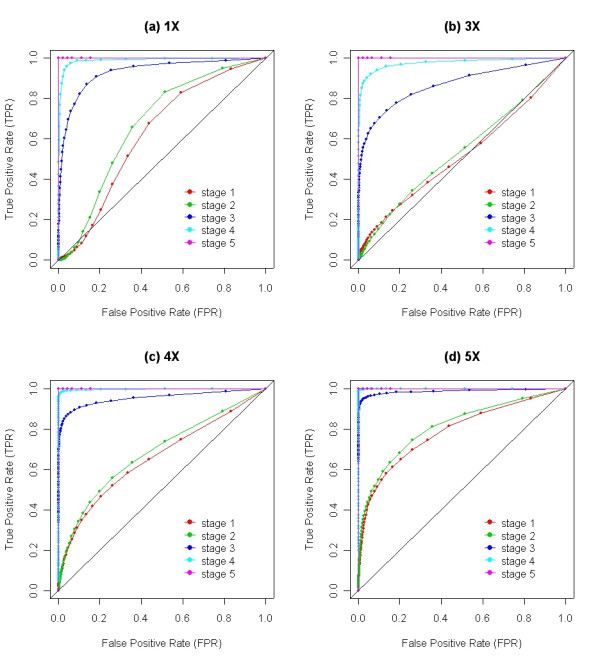
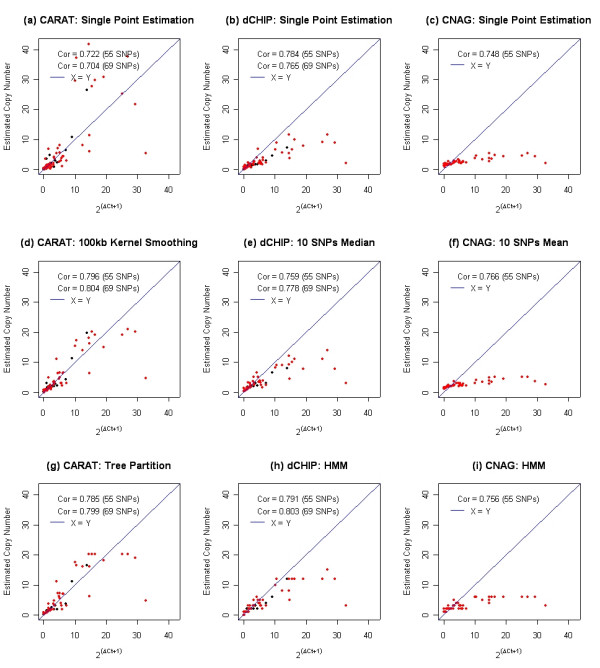
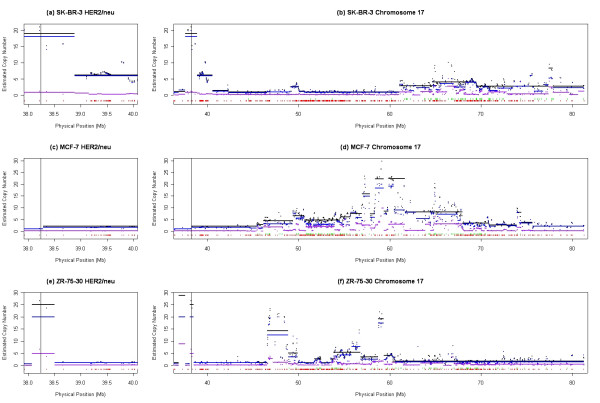
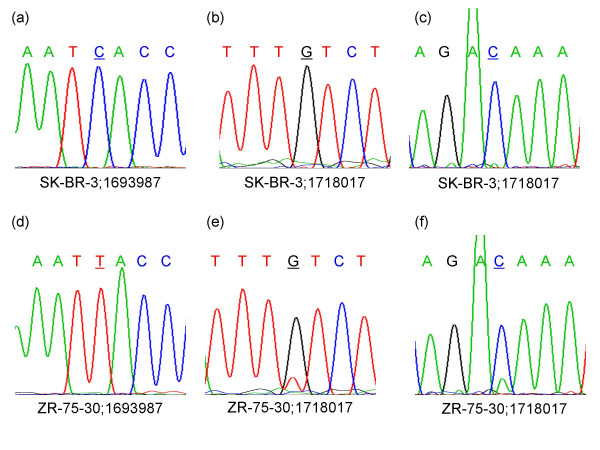
We describe a novel algorithm called CARAT (Copy Number Analysis with Regression And Tree) that uses probe intensity information to infer copy number in an allele-specific manner from high density DNA oligonuceotide arrays designed to genotype over 100,000 SNPs. Total and allele-specific copy number estimations using CARAT are independently evaluated for a subset of SNPs using quantitative PCR and allelic TaqMan reactions with several human breast cancer cell lines. The sensitivity and specificity of the algorithm are characterized using DNA samples containing differing numbers of X chromosomes as well as a test set of normal individuals. Results from the algorithm show a high degree of agreement with results from independent verification methods.
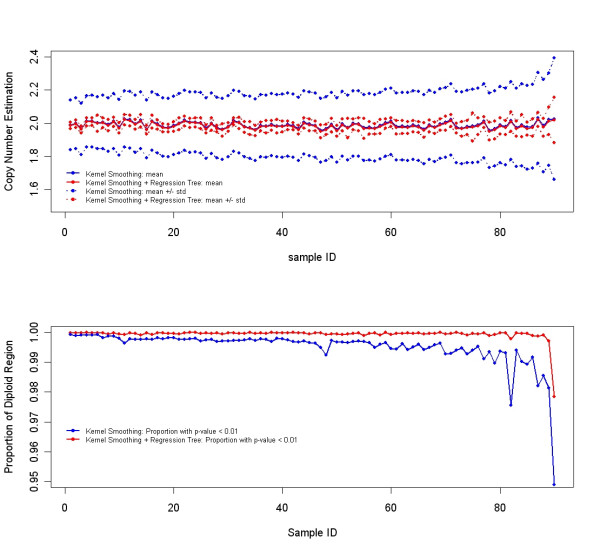
Overall, CARAT automatically detects regions with copy number variations and assigns a significance score to each alteration as well as generating allele-specific output. When coupled with SNP genotype calls from the same array, CARAT provides additional detail into the structure of genome wide alterations that can contribute to allelic imbalance.
DNA拷贝数改变是癌细胞核型的主要特征之一,可导致这些细胞出现复杂的表型。这些改变可导致细胞癌基因的增加以及肿瘤抑制基因的缺失,其范围可小至间隔区域,也可涉及整条染色体。准确检测这些变化的能力对于理解它们如何影响细胞生物学至关重要。
我们描述了一种名为CARAT(Copy Number Analysis with Regression And Tree,基于回归和树的拷贝数分析)的新算法,该算法利用探针强度信息,以等位基因特异性方式从旨在对超过100,000个单核苷酸多态性(SNP)进行基因分型的高密度DNA寡核苷酸阵列中推断拷贝数。使用定量PCR和等位基因TaqMan反应,对几种人类乳腺癌细胞系的一部分SNP独立评估使用CARAT进行的总拷贝数和等位基因特异性拷贝数估计。使用含有不同数量X染色体的DNA样本以及一组正常个体的测试集来表征该算法的敏感性和特异性。该算法的结果与独立验证方法的结果高度一致。
总体而言,CARAT可自动检测拷贝数变异区域,为每个改变赋予一个显著性分数,并生成等位基因特异性输出。当与来自同一阵列的SNP基因型调用相结合时,CARAT可提供全基因组改变结构的更多细节,这些改变可能导致等位基因失衡。