Bengtsson Henrik, Hössjer Ola
Mathematical Statistics, Centre for Mathematical Sciences, Lund University, Box 118, SE-221 00 Lund, Sweden.
BMC Bioinformatics. 2006 Mar 1;7:100. doi: 10.1186/1471-2105-7-100.
Low-level processing and normalization of microarray data are most important steps in microarray analysis, which have profound impact on downstream analysis. Multiple methods have been suggested to date, but it is not clear which is the best. It is therefore important to further study the different normalization methods in detail and the nature of microarray data in general.
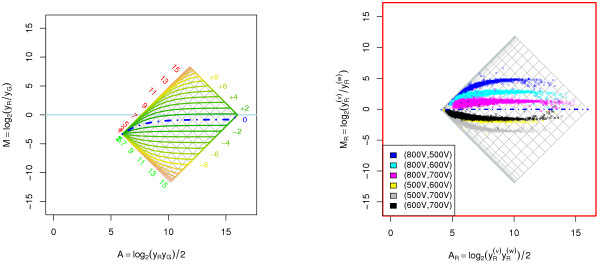
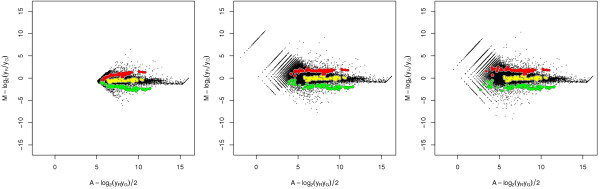
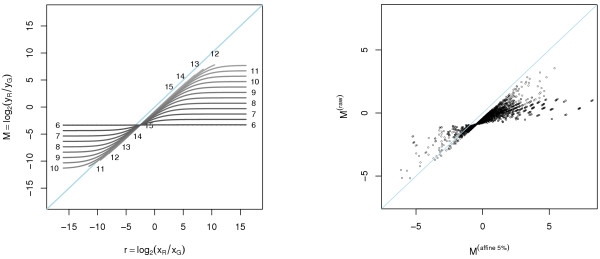
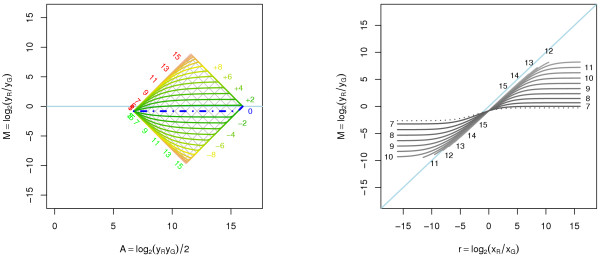
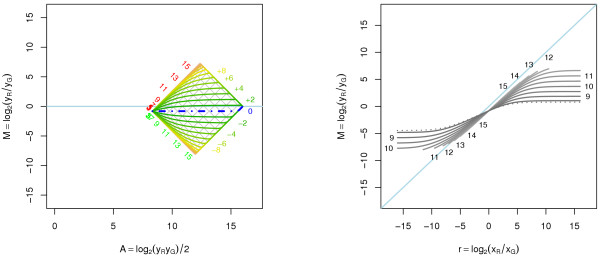
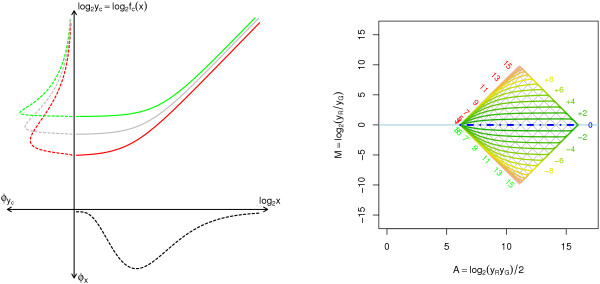
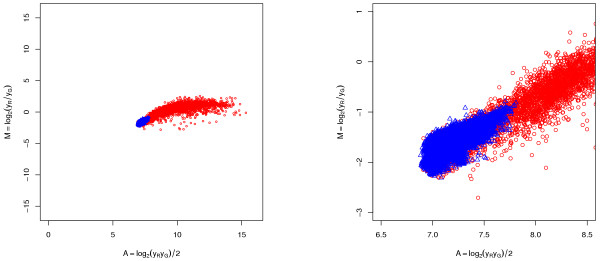
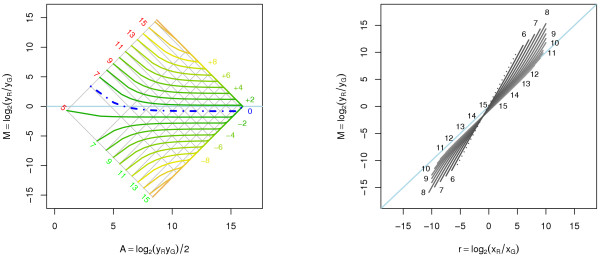
A methodological study of affine models for gene expression data is carried out. Focus is on two-channel comparative studies, but the findings generalize also to single- and multi-channel data. The discussion applies to spotted as well as in-situ synthesized microarray data. Existing normalization methods such as curve-fit ("lowess") normalization, parallel and perpendicular translation normalization, and quantile normalization, but also dye-swap normalization are revisited in the light of the affine model and their strengths and weaknesses are investigated in this context. As a direct result from this study, we propose a robust non-parametric multi-dimensional affine normalization method, which can be applied to any number of microarrays with any number of channels either individually or all at once. A high-quality cDNA microarray data set with spike-in controls is used to demonstrate the power of the affine model and the proposed normalization method.
We find that an affine model can explain non-linear intensity-dependent systematic effects in observed log-ratios. Affine normalization removes such artifacts for non-differentially expressed genes and assures that symmetry between negative and positive log-ratios is obtained, which is fundamental when identifying differentially expressed genes. In addition, affine normalization makes the empirical distributions in different channels more equal, which is the purpose of quantile normalization, and may also explain why dye-swap normalization works or fails. All methods are made available in the aroma package, which is a platform-independent package for R.
微阵列数据的低水平处理和标准化是微阵列分析中最重要的步骤,对下游分析有着深远影响。迄今为止已提出多种方法,但尚不清楚哪种方法最佳。因此,详细研究不同的标准化方法以及微阵列数据的一般性质非常重要。
开展了一项针对基因表达数据仿射模型的方法学研究。重点是双通道比较研究,但研究结果也适用于单通道和多通道数据。讨论适用于点阵式以及原位合成的微阵列数据。根据仿射模型重新审视了现有的标准化方法,如曲线拟合(“局部加权回归”)标准化、平行和垂直平移标准化、分位数标准化以及染料交换标准化,并在此背景下研究了它们的优缺点。作为本研究的直接成果,我们提出了一种稳健的非参数多维仿射标准化方法,该方法可单独或一次性应用于任意数量通道的任意数量微阵列。使用一个带有内参对照的高质量cDNA微阵列数据集来证明仿射模型和所提出的标准化方法的功效。
我们发现仿射模型可以解释观察到的对数比值中与强度相关的非线性系统效应。仿射标准化消除了非差异表达基因的此类假象,并确保获得正负对数比值之间的对称性,这在识别差异表达基因时至关重要。此外,仿射标准化使不同通道中的经验分布更加均等,这是分位数标准化的目的,也可能解释了染料交换标准化为何有效或无效。所有方法都可在aroma软件包中获取,该软件包是一个与平台无关的R软件包。