Keane Thomas M, Creevey Christopher J, Pentony Melissa M, Naughton Thomas J, Mclnerney James O
Department of Biology, National University of Ireland, Maynooth, Co. Kildare, Ireland.
BMC Evol Biol. 2006 Mar 24;6:29. doi: 10.1186/1471-2148-6-29.
In recent years, model based approaches such as maximum likelihood have become the methods of choice for constructing phylogenies. A number of authors have shown the importance of using adequate substitution models in order to produce accurate phylogenies. In the past, many empirical models of amino acid substitution have been derived using a variety of different methods and protein datasets. These matrices are normally used as surrogates, rather than deriving the maximum likelihood model from the dataset being examined. With few exceptions, selection between alternative matrices has been carried out in an ad hoc manner.
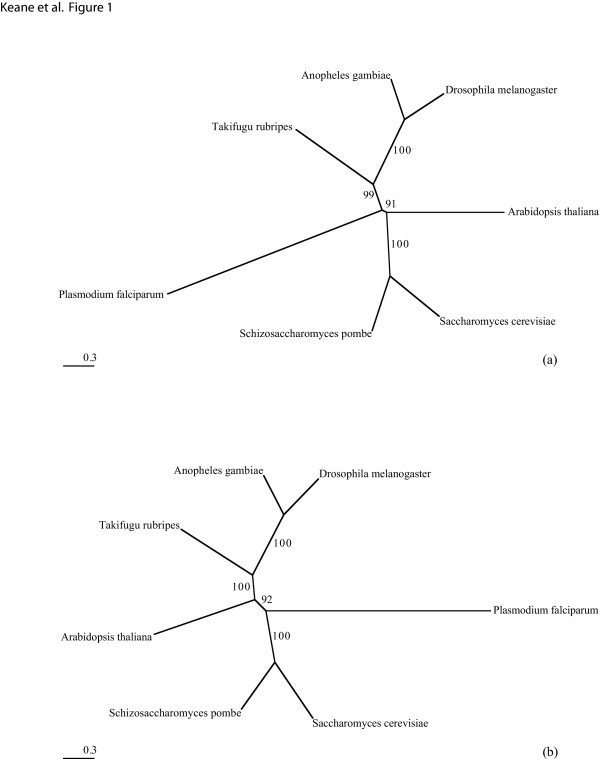
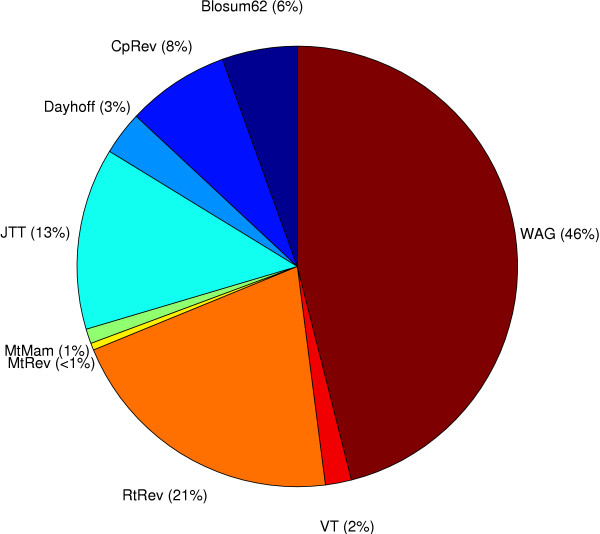
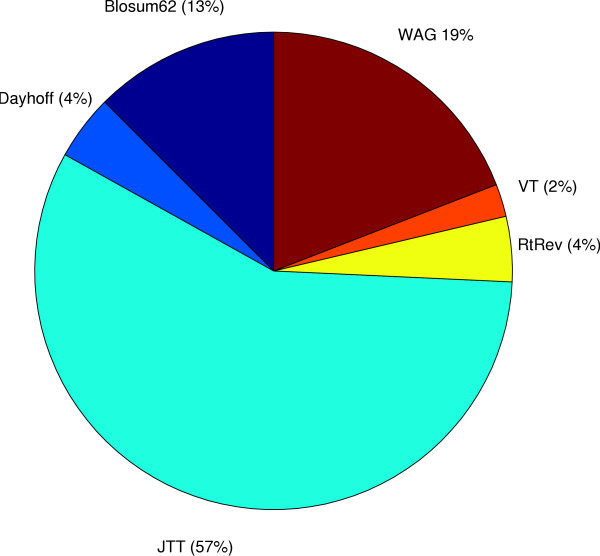
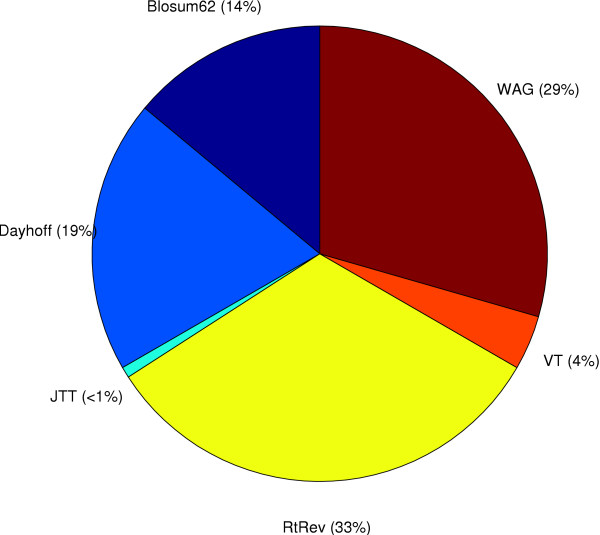
We start by highlighting the potential dangers of arbitrarily choosing protein models by demonstrating an empirical example where a single alignment can produce two topologically different and strongly supported phylogenies using two different arbitrarily-chosen amino acid substitution models. We demonstrate that in simple simulations, statistical methods of model selection are indeed robust and likely to be useful for protein model selection. We have investigated patterns of amino acid substitution among homologous sequences from the three Domains of life and our results show that no single amino acid matrix is optimal for any of the datasets. Perhaps most interestingly, we demonstrate that for two large datasets derived from the proteobacteria and archaea, one of the most favored models in both datasets is a model that was originally derived from retroviral Pol proteins.
This demonstrates that choosing protein models based on their source or method of construction may not be appropriate.
近年来,诸如最大似然法等基于模型的方法已成为构建系统发育树的首选方法。许多作者已表明使用适当的替换模型对于生成准确的系统发育树的重要性。过去,许多氨基酸替换的经验模型是使用各种不同方法和蛋白质数据集推导出来的。这些矩阵通常用作替代物,而不是从所研究的数据集中推导最大似然模型。除了少数例外,在替代矩阵之间的选择一直是以临时的方式进行的。
我们首先通过展示一个实证例子来突出任意选择蛋白质模型的潜在危险,在这个例子中,使用两种不同的任意选择的氨基酸替换模型,单个比对可以产生两个拓扑结构不同且得到有力支持的系统发育树。我们证明,在简单模拟中,模型选择的统计方法确实稳健,并且可能对蛋白质模型选择有用。我们研究了来自生命三个域的同源序列之间的氨基酸替换模式,我们的结果表明,对于任何数据集,没有单一的氨基酸矩阵是最优的。也许最有趣的是,我们证明,对于来自变形菌门和古菌的两个大型数据集,两个数据集中最受青睐的模型之一是最初从逆转录病毒Pol蛋白推导出来的模型。
这表明基于蛋白质模型的来源或构建方法来选择可能不合适。