Fan Wenhong, Khalid Najma, Hallahan Andrew R, Olson James M, Zhao Lue Ping
Division of Public Health Sciences, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N., Seattle, WA 98109, USA.
Theor Biol Med Model. 2006 Apr 7;3:19. doi: 10.1186/1742-4682-3-19.
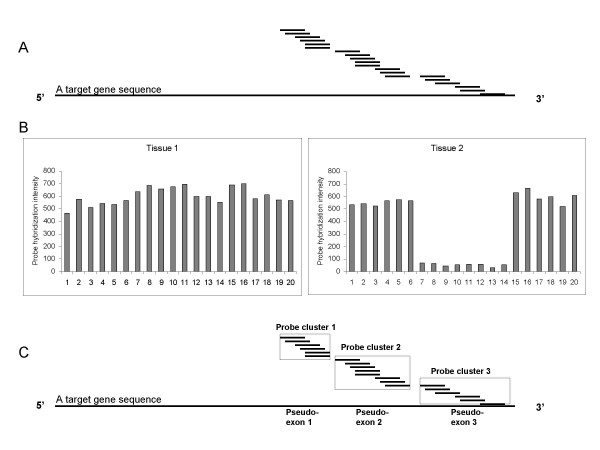
Alternative splicing of pre-messenger RNA results in RNA variants with combinations of selected exons. It is one of the essential biological functions and regulatory components in higher eukaryotic cells. Some of these variants are detectable with the Affymetrix GeneChip that uses multiple oligonucleotide probes (i.e. probe set), since the target sequences for the multiple probes are adjacent within each gene. Hybridization intensity from a probe correlates with abundance of the corresponding transcript. Although the multiple-probe feature in the current GeneChip was designed to assess expression values of individual genes, it also measures transcriptional abundance for a sub-region of a gene sequence. This additional capacity motivated us to develop a method to predict alternative splicing, taking advance of extensive repositories of GeneChip gene expression array data.
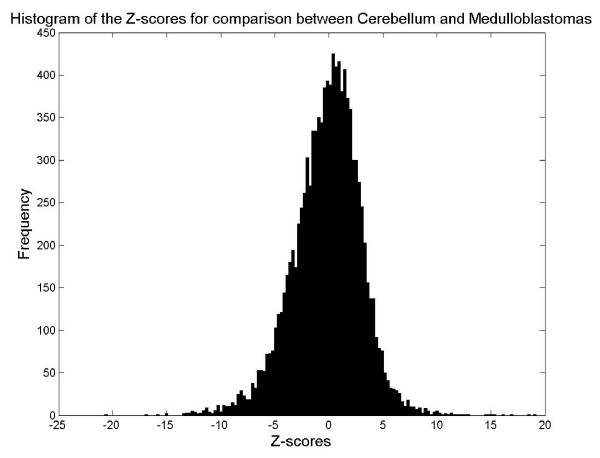
We developed a two-step approach to predict alternative splicing from GeneChip data. First, we clustered the probes from a probe set into pseudo-exons based on similarity of probe intensities and physical adjacency. A pseudo-exon is defined as a sequence in the gene within which multiple probes have comparable probe intensity values. Second, for each pseudo-exon, we assessed the statistical significance of the difference in probe intensity between two groups of samples. Differentially expressed pseudo-exons are predicted to be alternatively spliced. We applied our method to empirical data generated from GeneChip Hu6800 arrays, which include 7129 probe sets and twenty probes per probe set. The dataset consists of sixty-nine medulloblastoma (27 metastatic and 42 non-metastatic) samples and four cerebellum samples as normal controls. We predicted that 577 genes would be alternatively spliced when we compared normal cerebellum samples to medulloblastomas, and predicted that thirteen genes would be alternatively spliced when we compared metastatic medulloblastomas to non-metastatic ones. We checked the consistency of some of our findings with information in UCSC Human Genome Browser.
The two-step approach described in this paper is capable of predicting some alternative splicing from multiple oligonucleotide-based gene expression array data with GeneChip technology. Our method employs the extensive repositories of gene expression array data available and generates alternative splicing hypotheses, which can be further validated by experimental studies.
信使前体RNA的可变剪接产生具有选定外显子组合的RNA变体。它是高等真核细胞中基本的生物学功能和调控组成部分之一。使用多个寡核苷酸探针(即探针集)的Affymetrix基因芯片可检测到其中一些变体,因为多个探针的靶序列在每个基因内相邻。探针的杂交强度与相应转录本的丰度相关。尽管当前基因芯片中的多探针特性旨在评估单个基因的表达值,但它也可测量基因序列子区域的转录丰度。这种额外的能力促使我们利用基因芯片基因表达阵列数据的大量存储库开发一种预测可变剪接的方法。
我们开发了一种两步法从基因芯片数据预测可变剪接。首先,我们根据探针强度的相似性和物理邻接性将探针集中的探针聚类为假外显子。假外显子被定义为基因中的一个序列,其中多个探针具有可比的探针强度值。其次,对于每个假外显子,我们评估两组样本之间探针强度差异的统计显著性。差异表达的假外显子被预测为可变剪接。我们将我们的方法应用于从基因芯片Hu6800阵列生成的经验数据,该阵列包括7129个探针集,每个探针集有20个探针。数据集由69个髓母细胞瘤(27个转移性和42个非转移性)样本和4个小脑样本作为正常对照组成。当我们将正常小脑样本与髓母细胞瘤进行比较时,预测有577个基因会发生可变剪接;当我们将转移性髓母细胞瘤与非转移性髓母细胞瘤进行比较时,预测有13个基因会发生可变剪接。我们检查了我们的一些发现与UCSC人类基因组浏览器中的信息的一致性。
本文描述的两步法能够利用基因芯片技术从基于多个寡核苷酸的基因表达阵列数据中预测一些可变剪接。我们的方法利用了可用的基因表达阵列数据的大量存储库,并生成可变剪接假设,这些假设可通过实验研究进一步验证。