Seno Shigeto, Takenaka Yoichi, Kai Chikatoshi, Kawai Jun, Carninci Piero, Hayashizaki Yoshihide, Matsuda Hideo
Department of Bioinformatic Engineering, Graduate School of Information Science and Technology, Osaka University, Osaka, Japan.
PLoS Genet. 2006 Apr;2(4):e44. doi: 10.1371/journal.pgen.0020044. Epub 2006 Apr 28.
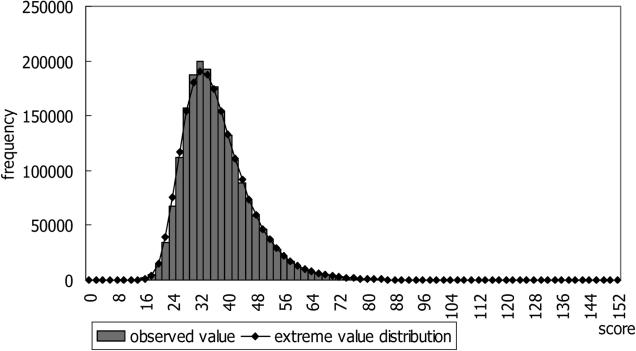
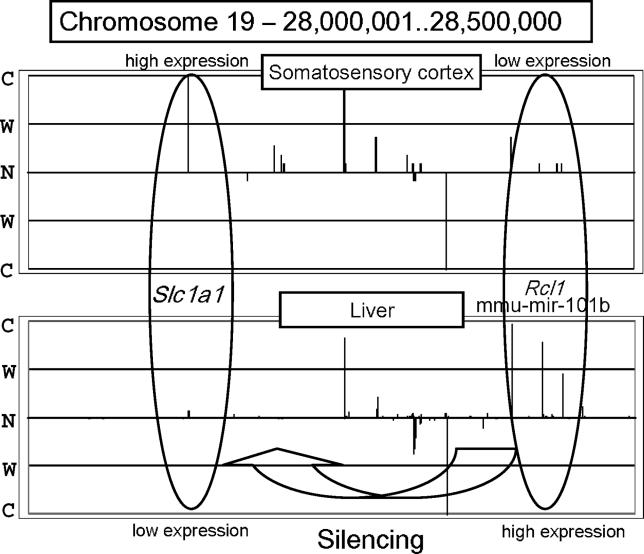
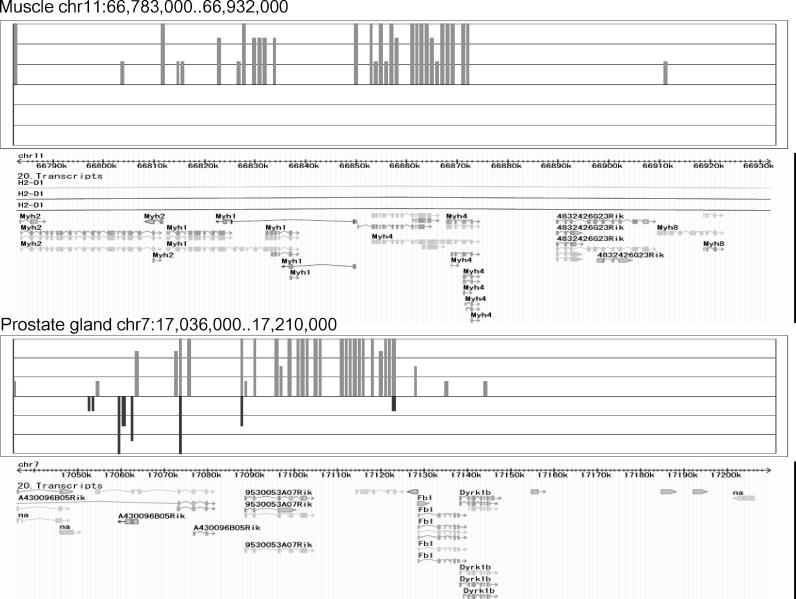
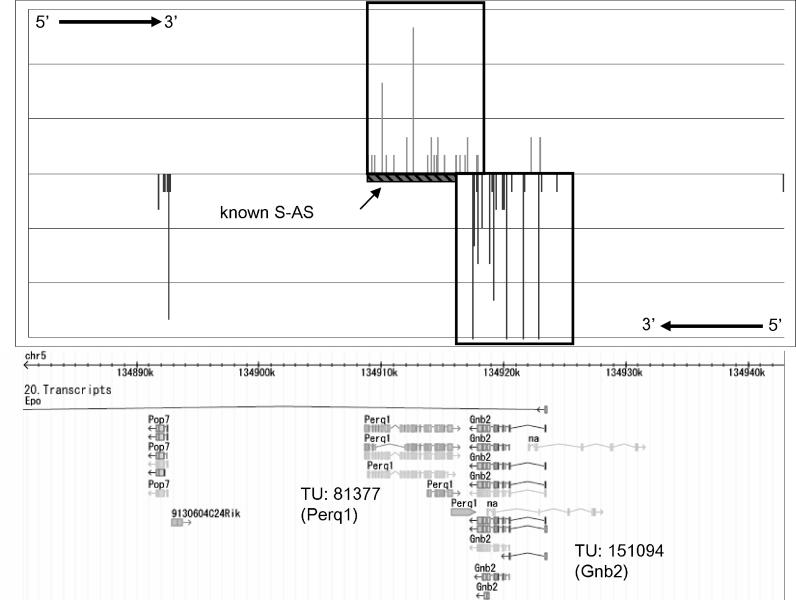
With the advancement of genome research, it is becoming clear that genes are not distributed on the genome in random order. Clusters of genes distributed at localized genome positions have been reported in several eukaryotes. Various correlations have been observed between the expressions of genes in adjacent or nearby positions along the chromosomes depending on tissue type and developmental stage. Moreover, in several cases, their transcripts, which control epigenetic transcription via processes such as transcriptional interference and genomic imprinting, occur in clusters. It is reasonable that genomic regions that have similar mechanisms show similar expression patterns and that the characteristics of expression in the same genomic regions differ depending on tissue type and developmental stage. In this study, we analyzed gene expression patterns using the cap analysis gene expression (CAGE) method for exploring systematic views of the mouse transcriptome. Counting the number of mapped CAGE tags for fixed-length regions allowed us to determine genomic expression levels. These expression levels were normalized, quantified, and converted into four types of descriptors, allowing the expression patterns along the genome to be represented by character strings. We analyzed them using dynamic programming in the same manner as for sequence analysis. We have developed a novel algorithm that provides a novel view of the genome from the perspective of genomic positional expression. In a similarity search of expression patterns across chromosomes and tissues, we found regions that had clusters of genes that showed expression patterns similar to each other depending on tissue type. Our results suggest the possibility that the regions that have sense-antisense transcription show similar expression patterns between forward and reverse strands.
随着基因组研究的进展,越来越清楚的是基因并非以随机顺序分布在基因组上。在几种真核生物中都报道了分布于局部基因组位置的基因簇。根据组织类型和发育阶段,沿着染色体在相邻或附近位置的基因表达之间观察到了各种相关性。此外,在一些情况下,它们通过转录干扰和基因组印记等过程控制表观遗传转录的转录本也成簇出现。具有相似机制的基因组区域显示出相似的表达模式,并且同一基因组区域的表达特征因组织类型和发育阶段而异,这是合理的。在本研究中,我们使用帽分析基因表达(CAGE)方法分析基因表达模式,以探索小鼠转录组的系统视图。对固定长度区域的映射CAGE标签数量进行计数使我们能够确定基因组表达水平。这些表达水平经过归一化、量化,并转换为四种类型的描述符,从而可以用字符串表示沿基因组的表达模式。我们以与序列分析相同的方式使用动态规划对其进行分析。我们开发了一种新算法,从基因组位置表达的角度提供了对基因组的全新视角。在跨染色体和组织的表达模式相似性搜索中,我们发现了一些区域,这些区域中的基因簇根据组织类型显示出彼此相似的表达模式。我们的结果表明,存在正义-反义转录的区域在正链和反链之间可能显示出相似的表达模式。