Yu Yiting, Kim H Stanley, Chua Hui Hoon, Lin Chi Ho, Sim Siew Hoon, Lin Daoxun, Derr Alan, Engels Reinhard, DeShazer David, Birren Bruce, Nierman William C, Tan Patrick
Genome Institute of Singapore, Singapore 138672, Republic of Singapore.
BMC Microbiol. 2006 May 26;6:46. doi: 10.1186/1471-2180-6-46.
The Gram-negative bacterium Burkholderia pseudomallei (Bp) is the causative agent of the human disease melioidosis. To understand the evolutionary mechanisms contributing to Bp virulence, we performed a comparative genomic analysis of Bp K96243 and B. thailandensis (Bt) E264, a closely related but avirulent relative.
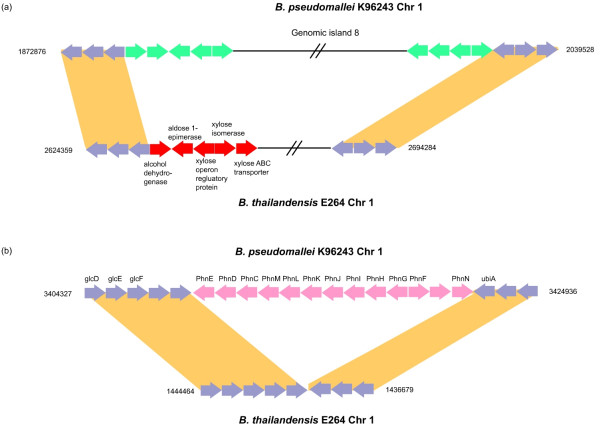
We found the Bp and Bt genomes to be broadly similar, comprising two highly syntenic chromosomes with comparable numbers of coding regions (CDs), protein family distributions, and horizontally acquired genomic islands, which we experimentally validated to be differentially present in multiple Bt isolates. By examining species-specific genomic regions, we derived molecular explanations for previously-known metabolic differences, discovered potentially new ones, and found that the acquisition of a capsular polysaccharide gene cluster in Bp, a key virulence component, is likely to have occurred non-randomly via replacement of an ancestral polysaccharide cluster. Virulence related genes, in particular members of the Type III secretion needle complex, were collectively more divergent between Bp and Bt compared to the rest of the genome, possibly contributing towards the ability of Bp to infect mammalian hosts. An analysis of pseudogenes between the two species revealed that protein inactivation events were significantly biased towards membrane-associated proteins in Bt and transcription factors in Bp.
Our results suggest that a limited number of horizontal-acquisition events, coupled with the fine-scale functional modulation of existing proteins, are likely to be the major drivers underlying Bp virulence. The extensive genomic similarity between Bp and Bt suggests that, in some cases, Bt could be used as a possible model system for studying certain aspects of Bp behavior.
革兰氏阴性菌类鼻疽伯克霍尔德菌(Bp)是人类类鼻疽病的病原体。为了解导致Bp毒力的进化机制,我们对Bp K96243和与其密切相关但无毒的泰国伯克霍尔德菌(Bt)E264进行了比较基因组分析。
我们发现Bp和Bt的基因组大致相似,由两条高度同线的染色体组成,编码区(CDs)数量、蛋白质家族分布以及水平获得的基因组岛数量相当,我们通过实验验证这些基因组岛在多个Bt分离株中存在差异。通过检查物种特异性基因组区域,我们对先前已知的代谢差异得出了分子解释,发现了潜在的新差异,并发现Bp中关键毒力成分荚膜多糖基因簇的获得可能是通过取代祖先多糖簇非随机发生的。与基因组的其他部分相比,Bp和Bt之间与毒力相关的基因,特别是III型分泌针复合物的成员,总体上差异更大,这可能有助于Bp感染哺乳动物宿主的能力。对这两个物种之间假基因的分析表明,蛋白质失活事件在Bt中明显偏向于膜相关蛋白,在Bp中则偏向于转录因子。
我们的结果表明,有限数量的水平获得事件,加上对现有蛋白质的精细功能调节,可能是Bp毒力的主要驱动因素。Bp和Bt之间广泛的基因组相似性表明,在某些情况下,Bt可以用作研究Bp行为某些方面的可能模型系统。