DeSantis T Z, Hugenholtz P, Keller K, Brodie E L, Larsen N, Piceno Y M, Phan R, Andersen G L
Lawrence Berkeley National Laboratory, Center for Environmental Biotechnology, Berkeley, CA, USA.
Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W394-9. doi: 10.1093/nar/gkl244.
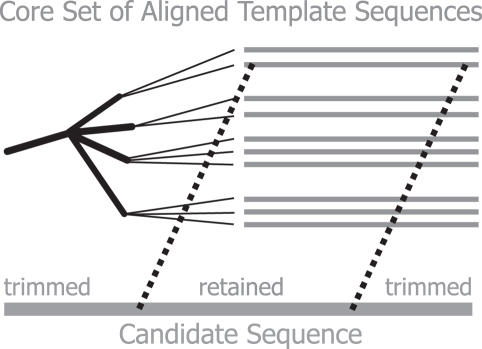
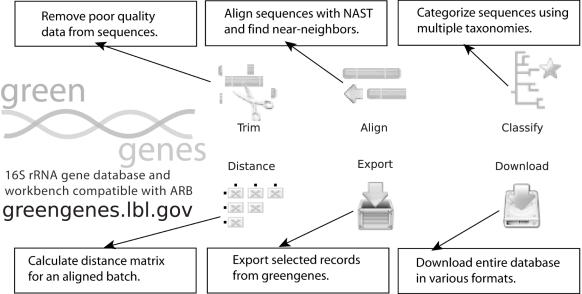
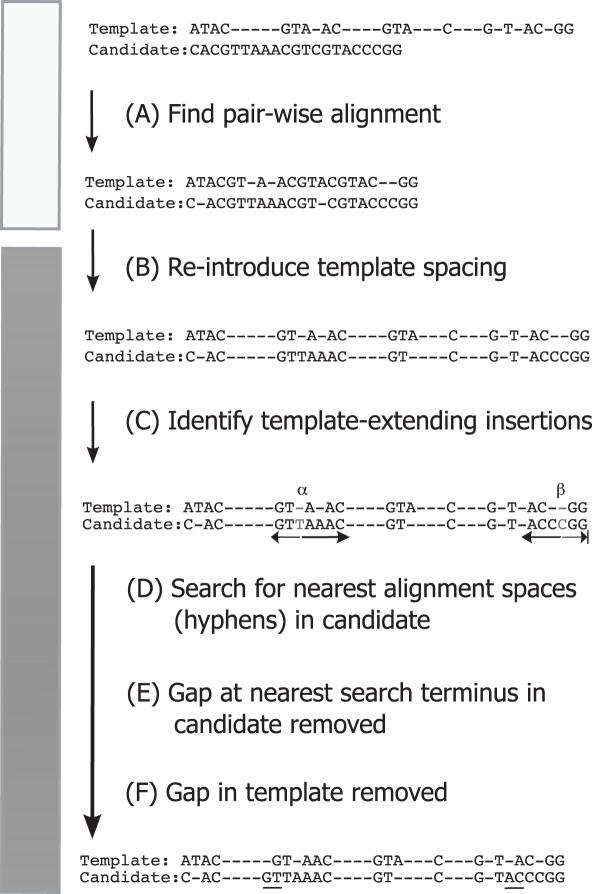
Microbiologists conducting surveys of bacterial and archaeal diversity often require comparative alignments of thousands of 16S rRNA genes collected from a sample. The computational resources and bioinformatics expertise required to construct such an alignment has inhibited high-throughput analysis. It was hypothesized that an online tool could be developed to efficiently align thousands of 16S rRNA genes via the NAST (Nearest Alignment Space Termination) algorithm for creating multiple sequence alignments (MSA). The tool was implemented with a web-interface at http://greengenes.lbl.gov/NAST. Each user-submitted sequence is compared with Greengenes' 'Core Set', comprising approximately 10,000 aligned non-chimeric sequences representative of the currently recognized diversity among bacteria and archaea. User sequences are oriented and paired with their closest match in the Core Set to serve as a template for inserting gap characters. Non-16S data (sequence from vector or surrounding genomic regions) are conveniently removed in the returned alignment. From the resulting MSA, distance matrices can be calculated for diversity estimates and organisms can be classified by taxonomy. The ability to align and categorize large sequence sets using a simple interface has enabled researchers with various experience levels to obtain bacterial and archaeal community profiles.
进行细菌和古菌多样性调查的微生物学家通常需要对从样本中收集的数千个16S rRNA基因进行比对。构建这样一个比对所需的计算资源和生物信息学专业知识阻碍了高通量分析。据推测,可以开发一种在线工具,通过NAST(最近比对空间终止)算法有效地比对数千个16S rRNA基因,以创建多序列比对(MSA)。该工具通过http://greengenes.lbl.gov/NAST的网络界面实现。每个用户提交的序列都与Greengenes的“核心集”进行比较,该核心集包含大约10,000个比对的非嵌合序列,代表了目前公认的细菌和古菌之间的多样性。用户序列被定向并与它们在核心集中最匹配的序列配对,作为插入空位字符的模板。在返回的比对中,可以方便地去除非16S数据(来自载体或周围基因组区域的序列)。从得到的MSA中,可以计算距离矩阵以进行多样性估计,并可以根据分类学对生物进行分类。使用简单界面比对和分类大型序列集的能力使具有不同经验水平的研究人员能够获得细菌和古菌群落概况。