Grau Jan, Ben-Gal Irad, Posch Stefan, Grosse Ivo
Institute of Computer Science, University Halle, 06099 Halle, Saale, Germany.
Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W529-33. doi: 10.1093/nar/gkl212.
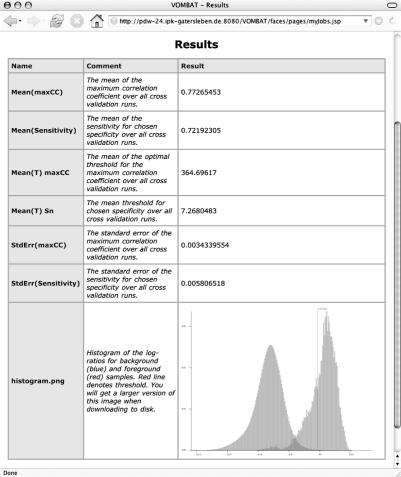
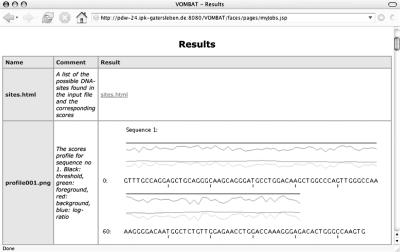
Variable order Markov models and variable order Bayesian trees have been proposed for the recognition of transcription factor binding sites, and it could be demonstrated that they outperform traditional models, such as position weight matrices, Markov models and Bayesian trees. We develop a web server for the recognition of DNA binding sites based on variable order Markov models and variable order Bayesian trees offering the following functionality: (i) given datasets with annotated binding sites and genomic background sequences, variable order Markov models and variable order Bayesian trees can be trained; (ii) given a set of trained models, putative DNA binding sites can be predicted in a given set of genomic sequences and (iii) given a dataset with annotated binding sites and a dataset with genomic background sequences, cross-validation experiments for different model combinations with different parameter settings can be performed. Several of the offered services are computationally demanding, such as genome-wide predictions of DNA binding sites in mammalian genomes or sets of 10(4)-fold cross-validation experiments for different model combinations based on problem-specific data sets. In order to execute these jobs, and in order to serve multiple users at the same time, the web server is attached to a Linux cluster with 150 processors. VOMBAT is available at http://pdw-24.ipk-gatersleben.de:8080/VOMBAT/.
可变阶马尔可夫模型和可变阶贝叶斯树已被用于识别转录因子结合位点,并且可以证明它们优于传统模型,如位置权重矩阵、马尔可夫模型和贝叶斯树。我们基于可变阶马尔可夫模型和可变阶贝叶斯树开发了一个用于识别DNA结合位点的网络服务器,它具有以下功能:(i)给定带有注释的结合位点和基因组背景序列的数据集,可以训练可变阶马尔可夫模型和可变阶贝叶斯树;(ii)给定一组训练好的模型,可以在给定的基因组序列集中预测潜在的DNA结合位点;(iii)给定一个带有注释的结合位点的数据集和一个带有基因组背景序列的数据集,可以针对不同参数设置的不同模型组合进行交叉验证实验。所提供的一些服务对计算要求很高,例如在哺乳动物基因组中进行全基因组范围的DNA结合位点预测,或基于特定问题数据集对不同模型组合进行10(4)倍交叉验证实验。为了执行这些任务,并为多个用户同时提供服务,该网络服务器连接到一个拥有150个处理器的Linux集群。VOMBAT可在http://pdw-24.ipk-gatersleben.de:8080/VOMBAT/获取。